Over the years, we have worked with scientists from UNIL, EPFL, CHUV and some other institutions, and are co-authors on over 60 publications, covering a wide range of topics (human diseases, genome assembly, chromosomal organization, environment and evolution, bacterial resistance—to name a few).
Posters
A broad range of expertise to advance your projects (2025)
A broad range of expertise to advance your projects (2025)
Design and development of web applications (2025)
Image Analysis (2026)
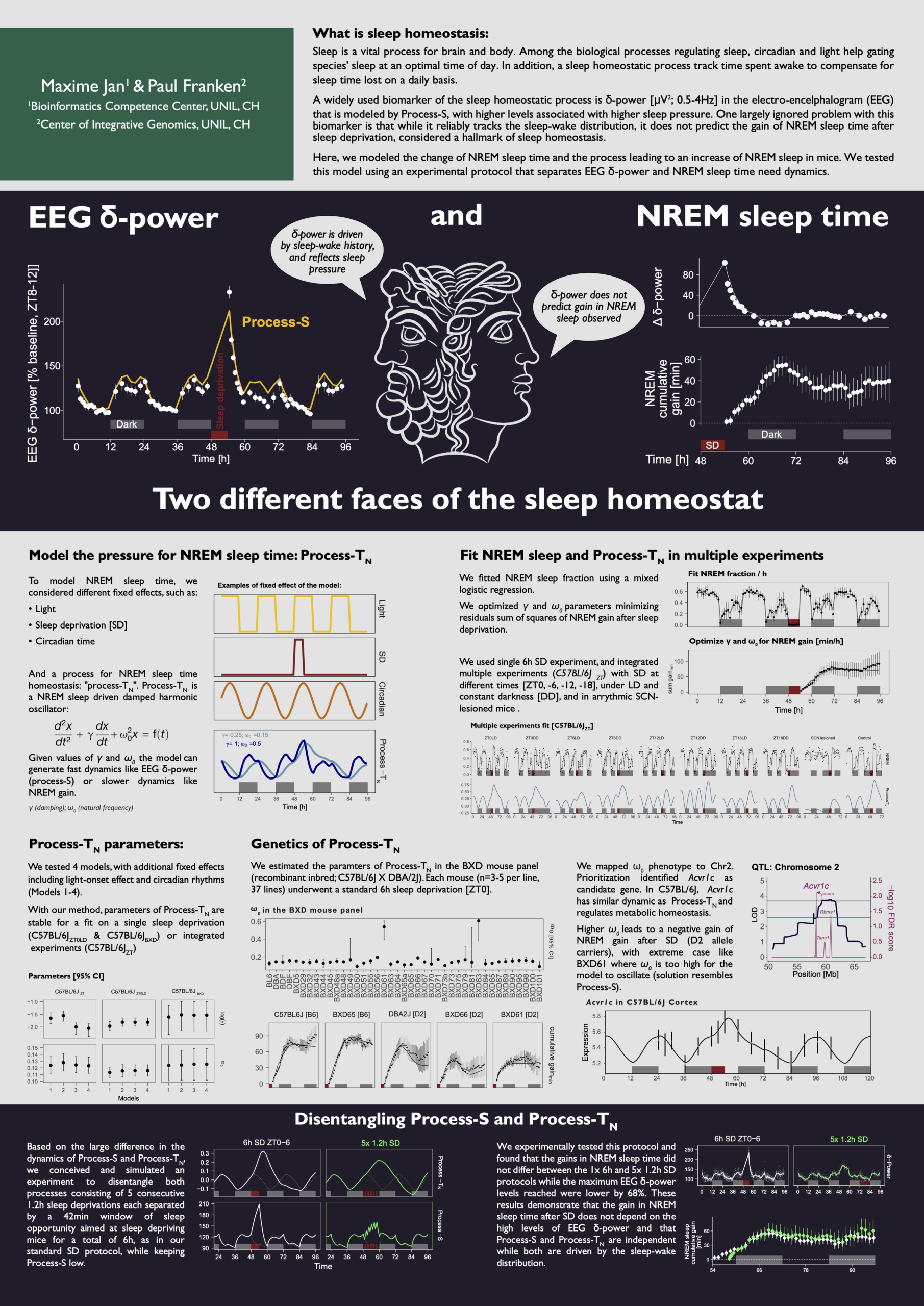
Two different faces of the sleep homeostat (2026)
Articles
id
Authors
authors
Title
Year
Journal
pmid
created_at
doi
published_at
first_author
journal_id
user_id
Volume
issue
abstract
Link to paper
27
C. Sala, A. Benjak, D. Goletti, S. Banu, J. Mazza-Stadler, K. Jaton, P. Busso, S. Remm, M. Leleu, J. Rougemont, F. Palmieri, G. Cuzzi, O. Butera, V. Vanini, S. Kabir, S.M.M. Rahman, L. Nicod, S.T. Cole
C Sala et al.
Multicenter analysis of sputum microbiota in tuberculosis patients.
2020
PloS one
33,044,973
10/12/2020 09:38
01/01/2020 00:00
Sala
6
15
10
The impact of tuberculosis and of anti-tuberculosis therapy on composition and modification of human lung microbiota has been the object of several investigations. However, no clear outcome has been presented so far and the relationship between M. tuberculosis pulmonary infection and the resident lung microbiota remains vague. In this work we describe the results obtained from a multicenter study of the microbiota of sputum samples from patients with tuberculosis or unrelated lung diseases and healthy donors recruited in Switzerland, Italy and Bangladesh, with the ultimate goal of discovering a microbiota-based biomarker associated with tuberculosis. Bacterial 16S rDNA amplification, high-throughput sequencing and extensive bioinformatic analyses revealed patient-specific flora and high variability in taxon abundance. No common signature could be identified among the individuals enrolled except for minor differences which were not consistent among the different geographical settings. Moreover, anti-tuberculosis therapy did not cause any important variation in microbiota diversity, thus precluding its exploitation as a biomarker for the follow up of tuberculosis patients undergoing treatment.
28
H.K. Mod, D. Scherrer, V. Di Cola, O. Broennimann, Q. Blandenier, F.T. Breiner, A. Buri, J. Goudet, N. Guex, E. Lara, E.A.D. Mitchell, H. Niculita-Hirzel, M. Pagni, L. Pellissier, E. Pinto-Figueroa, I.R. Sanders, B.R. Schmidt, C.V.W. Seppey, D. Singer, S. Ursenbacher, E. Yashiro, J.R. van der Meer, A. Guisan
HK Mod et al.
Greater topoclimatic control of above- versus below-ground communities.
2020
Global change biology
32,866,994
10/12/2020 09:38
01/12/2020 00:00
Mod
21
26
12
Assessing the degree to which climate explains the spatial distributions of different taxonomic and functional groups is essential for anticipating the effects of climate change on ecosystems. Most effort so far has focused on above-ground organisms, which offer only a partial view on the response of biodiversity to environmental gradients. Here including both above- and below-ground organisms, we quantified the degree of topoclimatic control on the occurrence patterns of >1,500 taxa and phylotypes along a c. 3,000 m elevation gradient, by fitting species distribution models. Higher model performances for animals and plants than for soil microbes (fungi, bacteria and protists) suggest that the direct influence of topoclimate is stronger on above-ground species than on below-ground microorganisms. Accordingly, direct climate change effects are predicted to be stronger for above-ground than for below-ground taxa, whereas factors expressing local soil microclimate and geochemistry are likely more important to explain and forecast the occurrence patterns of soil microbiota. Detailed mapping and future scenarios of soil microclimate and microhabitats, together with comparative studies of interacting and ecologically dependent above- and below-ground biota, are thus needed to understand and realistically forecast the future distribution of ecosystems.
29
A. Gleizes, F. Laubscher, N. Guex, C. Iseli, T. Junier, S. Cordey, J. Fellay, I. Xenarios, L. Kaiser, P.L. Mercier
A Gleizes et al.
Virosaurus A Reference to Explore and Capture Virus Genetic Diversity.
2020
Viruses
33,139,591
10/12/2020 09:38
01/11/2020 00:00
Gleizes
22
12
11
The huge genetic diversity of circulating viruses is a challenge for diagnostic assays for emerging or rare viral diseases. High-throughput technology offers a new opportunity to explore the global virome of patients without preconception about the culpable pathogens. It requires a solid reference dataset to be accurate. Virosaurus has been designed to offer a non-biased, automatized and annotated database for clinical metagenomics studies and diagnosis. Raw viral sequences have been extracted from GenBank, and cleaned up to remove potentially erroneous sequences. Complete sequences have been identified for all genera infecting vertebrates, plants and other eukaryotes (insect, fungus, etc.). To facilitate the analysis of clinically relevant viruses, we have annotated all sequences with official and common virus names, acronym, genotypes, and genomic features (linear, circular, DNA, RNA, etc.). Sequences have been clustered to remove redundancy at 90% or 98% identity. The analysis of clustering results reveals the state of the virus genetic landscape knowledge. Because herpes and poxviruses were under-represented in complete genomes considering their potential diversity in nature, we used genes instead of complete genomes for those in Virosaurus.
30
T. Caputo, V.D.T. Tran, N. Bararpour, C. Winkler, G. Aguileta, K.B. Trang, G.M.P. Giordano Attianese, A. Wilson, A. Thomas, M. Pagni, N. Guex, B. Desvergne, F. Gilardi
T Caputo et al.
Anti-adipogenic signals at the onset of obesity-related inflammation in white adipose tissue.
2021
Cellular and molecular life sciences : CMLS
32,157,317
10/12/2020 09:39
01/01/2021 00:00
Caputo
23
78
1
Chronic inflammation that affects primarily metabolic organs, such as white adipose tissue (WAT), is considered as a major cause of human obesity-associated co-morbidities. However, the molecular mechanisms initiating this inflammation in WAT are poorly understood. By combining transcriptomics, ChIP-seq and modeling approaches, we studied the global early and late responses to a high-fat diet (HFD) in visceral (vWAT) and subcutaneous (scWAT) AT, the first being more prone to obesity-induced inflammation. HFD rapidly triggers proliferation of adipocyte precursors within vWAT. However, concomitant antiadipogenic signals limit vWAT hyperplastic expansion by interfering with the differentiation of proliferating adipocyte precursors. Conversely, in scWAT, residing beige adipocytes lose their oxidizing properties and allow storage of excessive fatty acids. This phase is followed by tissue hyperplastic growth and increased angiogenic signals, which further enable scWAT expansion without generating inflammation. Our data indicate that scWAT and vWAT differential ability to modulate adipocyte number and differentiation in response to obesogenic stimuli has a crucial impact on the different susceptibility to obesity-related inflammation of these adipose tissue depots.
26
F.P.A. David, M. Litovchenko, B. Deplancke, V. Gardeux
FPA David et al.
ASAP 2020 update: an open, scalable and interactive web-based portal for (single-cell) omics analyses.
2020
Nucleic acids research
32,449,934
09/12/2020 17:34
02/07/2020 00:00
David
4
1
48
W1
Single-cell omics enables researchers to dissect biological systems at a resolution that was unthinkable just 10 years ago. However, this analytical revolution also triggered new demands in 'big data' management, forcing researchers to stay up to speed with increasingly complex analytical processes and rapidly evolving methods. To render these processes and approaches more accessible, we developed the web-based, collaborative portal ASAP (Automated Single-cell Analysis Portal). Our primary goal is thereby to democratize single-cell omics data analyses (scRNA-seq and more recently scATAC-seq). By taking advantage of a Docker system to enhance reproducibility, and novel bioinformatics approaches that were recently developed for improving scalability, ASAP meets challenging requirements set by recent cell atlasing efforts such as the Human (HCA) and Fly (FCA) Cell Atlas Projects. Specifically, ASAP can now handle datasets containing millions of cells, integrating intuitive tools that allow researchers to collaborate on the same project synchronously. ASAP tools are versioned, and researchers can create unique access IDs for storing complete analyses that can be reproduced or completed by others. Finally, ASAP does not require any installation and provides a full and modular single-cell RNA-seq analysis pipeline. ASAP is freely available at https://asap.epfl.ch.
31
P. De Nittis, S. Efthymiou, A. Sarre, N. Guex, J. Chrast, A. Putoux, T. Sultan, J. Raza Alvi, Z. Ur Rahman, F. Zafar, N. Rana, F. Rahman, N. Anwar, S. Maqbool, M.S. Zaki, J.G. Gleeson, D. Murphy, H. Galehdari, G. Shariati, N. Mazaheri, A. Sedaghat, , G. Lesca, N. Chatron, V. Salpietro, M. Christoforou, H. Houlden, W.F. Simonds, T. Pedrazzini, R. Maroofian, A. Reymond
P De Nittis et al.
Inhibition of G-protein signalling in cardiac dysfunction of intellectual developmental disorder with cardiac arrhythmia (IDDCA) syndrome.
2021
Journal of medical genetics
33,172,956
10/12/2020 09:39
01/12/2021 00:00
De Nittis
24
58
12
Pathogenic variants of GNB5 encoding the β5 subunit of the guanine nucleotide-binding protein cause IDDCA syndrome, an autosomal recessive neurodevelopmental disorder associated with cognitive disability and cardiac arrhythmia, particularly severe bradycardia.
32
J.T. Hannich, A.G. Haribowo, S. Gentina, M. Paillard, L. Gomez, B. Pillot, H. Thibault, D. Abegg, N. Guex, A. Zumbuehl, A. Adibekian, M. Ovize, J.C. Martinou, H. Riezman
JT Hannich et al.
1-Deoxydihydroceramide causes anoxic death by impairing chaperonin-mediated protein folding.
2019
Nature metabolism
32,694,842
16/12/2020 09:04
01/10/2019 00:00
Hannich
70
1
10
Ischaemic heart disease and stroke are the most common causes of death worldwide. Anoxia, defined as the lack of oxygen, is commonly seen in both these pathologies and triggers profound metabolic and cellular changes. Sphingolipids have been implicated in anoxia injury, but the pathomechanism is unknown. Here we show that anoxia-associated injury causes accumulation of the non-canonical sphingolipid 1-deoxydihydroceramide (DoxDHCer). Anoxia causes an imbalance between serine and alanine resulting in a switch from normal serine-derived sphinganine biosynthesis to non-canonical alanine-derived 1-deoxysphinganine. 1-Deoxysphinganine is incorporated into DoxDHCer, which impairs actin folding via the cytosolic chaperonin TRiC, leading to growth arrest in yeast, increased cell death upon anoxia-reoxygenation in worms and ischaemia-reperfusion injury in mouse hearts. Prevention of DoxDHCer accumulation in worms and in mouse hearts resulted in decreased anoxia-induced injury. These findings unravel key metabolic changes during oxygen deprivation and point to novel strategies to avoid tissue damage and death.
43
B.D. Weger, C. Gobet, F.P.A. David, F. Atger, E. Martin, N.E. Phillips, A. Charpagne, M. Weger, F. Naef, F. Gachon
BD Weger et al.
Systematic analysis of differential rhythmic liver gene expression mediated by the circadian clock and feeding rhythms.
2021
Proceedings of the National Academy of Sciences of the United States of America
33,452,134
17/01/2021 11:35
19/01/2021 00:00
Weger
14
118
3
The circadian clock and feeding rhythms are both important regulators of rhythmic gene expression in the liver. To further dissect the respective contributions of feeding and the clock, we analyzed differential rhythmicity of liver tissue samples across several conditions. We developed a statistical method tailored to compare rhythmic liver messenger RNA (mRNA) expression in mouse knockout models of multiple clock genes, as well as PARbZip output transcription factors (Hlf/Dbp/Tef). Mice were exposed to ad libitum or night-restricted feeding under regular light-dark cycles. During ad libitum feeding, genetic ablation of the core clock attenuated rhythmic-feeding patterns, which could be restored by the night-restricted feeding regimen. High-amplitude mRNA expression rhythms in wild-type livers were driven by the circadian clock, but rhythmic feeding also contributed to rhythmic gene expression, albeit with significantly lower amplitudes. We observed that Bmal1 and Cry1/2 knockouts differed in their residual rhythmic gene expression. Differences in mean expression levels between wild types and knockouts correlated with rhythmic gene expression in wild type. Surprisingly, in PARbZip knockout mice, the mean expression levels of PARbZip targets were more strongly impacted than their rhythms, potentially due to the rhythmic activity of the D-box-repressor NFIL3. Genes that lost rhythmicity in PARbZip knockouts were identified to be indirect targets. Our findings provide insights into the diurnal transcriptome in mouse liver as we identified the differential contributions of several core clock regulators. In addition, we gained more insights on the specific effects of the feeding-fasting cycle.
44
J. den Hoed, E. de Boer, N. Voisin, A.J.M. Dingemans, N. Guex, L. Wiel, C. Nellaker, S.M. Amudhavalli, S. Banka, F.S. Bena, B. Ben-Zeev, V.R. Bonagura, A.L. Bruel, T. Brunet, H.G. Brunner, H.B. Chew, J. Chrast, L. Cimbalistienė, H. Coon, , E.C. Délot, F. Démurger, A.S. Denommé-Pichon, C. Depienne, D. Donnai, D.A. Dyment, O. Elpeleg, L. Faivre, C. Gilissen, L. Granger, B. Haber, Y. Hachiya, Y.H. Abedi, J. Hanebeck, J.Y. Hehir-Kwa, B. Horist, T. Itai, A. Jackson, R. Jewell, K.L. Jones, S. Joss, H. Kashii, M. Kato, A.A. Kattentidt-Mouravieva, F. Kok, U. Kotzaeridou, V. Krishnamurthy, V. Kučinskas, A. Kuechler, A. Lavillaureix, P. Liu, L. Manwaring, N. Matsumoto, B. Mazel, K. McWalter, V. Meiner, M.A. Mikati, S. Miyatake, T. Mizuguchi, L.H. Moey, S. Mohammed, H. Mor-Shaked, H. Mountford, R. Newbury-Ecob, S. Odent, L. Orec, M. Osmond, T.B. Palculict, M. Parker, A.K. Petersen, R. Pfundt, E. Preikšaitienė, K. Radtke, E. Ranza, J.A. Rosenfeld, T. Santiago-Sim, C. Schwager, M. Sinnema, L. Snijders Blok, R.C. Spillmann, A.P.A. Stegmann, I. Thiffault, L. Tran, A. Vaknin-Dembinsky, J.H. Vedovato-Dos-Santos, S.A. Schrier Vergano, E. Vilain, A. Vitobello, M. Wagner, A. Waheeb, M. Willing, B. Zuccarelli, U. Kini, D.F. Newbury, T. Kleefstra, A. Reymond, S.E. Fisher, L.E.L.M. Vissers
J den Hoed et al.
Mutation-specific pathophysiological mechanisms define different neurodevelopmental disorders associated with SATB1 dysfunction.
2021
American journal of human genetics
33,513,338
30/01/2021 11:35
04/02/2021 00:00
den Hoed
399
108
2
Whereas large-scale statistical analyses can robustly identify disease-gene relationships, they do not accurately capture genotype-phenotype correlations or disease mechanisms. We use multiple lines of independent evidence to show that different variant types in a single gene, SATB1, cause clinically overlapping but distinct neurodevelopmental disorders. Clinical evaluation of 42 individuals carrying SATB1 variants identified overt genotype-phenotype relationships, associated with different pathophysiological mechanisms, established by functional assays. Missense variants in the CUT1 and CUT2 DNA-binding domains result in stronger chromatin binding, increased transcriptional repression, and a severe phenotype. In contrast, variants predicted to result in haploinsufficiency are associated with a milder clinical presentation. A similarly mild phenotype is observed for individuals with premature protein truncating variants that escape nonsense-mediated decay, which are transcriptionally active but mislocalized in the cell. Our results suggest that in-depth mutation-specific genotype-phenotype studies are essential to capture full disease complexity and to explain phenotypic variability.
45
A. Kaushal, G. Mohana, J. Dorier, I. Özdemir, A. Omer, P. Cousin, A. Semenova, M. Taschner, O. Dergai, F. Marzetta, C. Iseli, Y. Eliaz, D. Weisz, M.S. Shamim, N. Guex, E. Lieberman Aiden, M.C. Gambetta
A Kaushal et al.
CTCF loss has limited effects on global genome architecture in Drosophila despite critical regulatory functions.
2021
Nature communications
33,579,945
14/02/2021 11:35
12/02/2021 00:00
Kaushal
60
12
1
Vertebrate genomes are partitioned into contact domains defined by enhanced internal contact frequency and formed by two principal mechanisms: compartmentalization of transcriptionally active and inactive domains, and stalling of chromosomal loop-extruding cohesin by CTCF bound at domain boundaries. While Drosophila has widespread contact domains and CTCF, it is currently unclear whether CTCF-dependent domains exist in flies. We genetically ablate CTCF in Drosophila and examine impacts on genome folding and transcriptional regulation in the central nervous system. We find that CTCF is required to form a small fraction of all domain boundaries, while critically controlling expression patterns of certain genes and supporting nervous system function. We also find that CTCF recruits the pervasive boundary-associated factor Cp190 to CTCF-occupied boundaries and co-regulates a subset of genes near boundaries together with Cp190. These results highlight a profound difference in CTCF-requirement for genome folding in flies and vertebrates, in which a large fraction of boundaries are CTCF-dependent and suggest that CTCF has played mutable roles in genome architecture and direct gene expression control during metazoan evolution.
46
N. Bararpour, F. Gilardi, C. Carmeli, J. Sidibe, J. Ivanisevic, T. Caputo, M. Augsburger, S. Grabherr, B. Desvergne, N. Guex, M. Bochud, A. Thomas
N Bararpour et al.
DBnorm as an R package for the comparison and selection of appropriate statistical methods for batch effect correction in metabolomic studies.
2021
Scientific reports
33,707,505
13/03/2021 11:35
11/03/2021 00:00
Bararpour
81
11
1
As a powerful phenotyping technology, metabolomics provides new opportunities in biomarker discovery through metabolome-wide association studies (MWAS) and the identification of metabolites having a regulatory effect in various biological processes. While mass spectrometry-based (MS) metabolomics assays are endowed with high throughput and sensitivity, MWAS are doomed to long-term data acquisition generating an overtime-analytical signal drift that can hinder the uncovering of real biologically relevant changes. We developed "dbnorm", a package in the R environment, which allows for an easy comparison of the model performance of advanced statistical tools commonly used in metabolomics to remove batch effects from large metabolomics datasets. "dbnorm" integrates advanced statistical tools to inspect the dataset structure not only at the macroscopic (sample batches) scale, but also at the microscopic (metabolic features) level. To compare the model performance on data correction, "dbnorm" assigns a score that help users identify the best fitting model for each dataset. In this study, we applied "dbnorm" to two large-scale metabolomics datasets as a proof of concept. We demonstrate that "dbnorm" allows for the accurate selection of the most appropriate statistical tool to efficiently remove the overtime signal drift and to focus on the relevant biological components of complex datasets.
48
H.K. Mod, A. Buri, E. Yashiro, N. Guex, L. Malard, E. Pinto-Figueroa, M. Pagni, H. Niculita-Hirzel, J.R. van der Meer, A. Guisan
HK Mod et al.
Predicting spatial patterns of soil bacteria under current and future environmental conditions.
2021
The ISME journal
33,712,699
14/03/2021 11:35
01/09/2021 00:00
Mod
133
15
9
Soil bacteria are largely missing from future biodiversity assessments hindering comprehensive forecasts of ecosystem changes. Soil bacterial communities are expected to be more strongly driven by pH and less by other edaphic and climatic factors. Thus, alkalinisation or acidification along with climate change may influence soil bacteria, with subsequent influences for example on nutrient cycling and vegetation. Future forecasts of soil bacteria are therefore needed. We applied species distribution modelling (SDM) to quantify the roles of environmental factors in governing spatial abundance distribution of soil bacterial OTUs and to predict how future changes in these factors may change bacterial communities in a temperate mountain area. Models indicated that factors related to soil (especially pH), climate and/or topography explain and predict part of the abundance distribution of most OTUs. This supports the expectations that microorganisms have specific environmental requirements (i.e., niches/envelopes) and that they should accordingly respond to environmental changes. Our predictions indicate a stronger role of pH over other predictors (e.g. climate) in governing distributions of bacteria, yet the predicted future changes in bacteria communities are smaller than their current variation across space. The extent of bacterial community change predictions varies as a function of elevation, but in general, deviations from neutral soil pH are expected to decrease abundances and diversity of bacteria. Our findings highlight the need to account for edaphic changes, along with climate changes, in future forecasts of soil bacteria.
50
H.H. Schede, C.G. Schneider, J. Stergiadou, L.E. Borm, A. Ranjak, T.M. Yamawaki, F.P.A. David, P. Lönnerberg, M.A. Tosches, S. Codeluppi, G. La Manno
HH Schede et al.
Spatial tissue profiling by imaging-free molecular tomography.
2021
Nature biotechnology
33,875,865
22/04/2021 10:35
01/08/2021 00:00
Schede
400
39
8
Several techniques are currently being developed for spatially resolved omics profiling, but each new method requires the setup of specific detection strategies or specialized instrumentation. Here we describe an imaging-free framework to localize high-throughput readouts within a tissue by cutting the sample into thin strips in a way that allows subsequent image reconstruction. We implemented this framework to transform a low-input RNA sequencing protocol into an imaging-free spatial transcriptomics technique (called STRP-seq) and validated it by profiling the spatial transcriptome of the mouse brain. We applied the technique to the brain of the Australian bearded dragon, Pogona vitticeps. Our results reveal the molecular anatomy of the telencephalon of this lizard, providing evidence for a marked regionalization of the reptilian pallium and subpallium. We expect that STRP-seq can be used to derive spatially resolved data from a range of other omics techniques.
51
S. Cordey, F. Laubscher, M.A. Hartley, T. Junier, K. Keitel, M. Docquier, N. Guex, C. Iseli, G. Vieille, P. Le Mercier, A. Gleizes, J. Samaka, T. Mlaganile, F. Kagoro, J. Masimba, Z. Said, H. Temba, G.H. Elbanna, C. Tapparel, M.C. Zanella, I. Xenarios, J. Fellay, V. D'Acremont, L. Kaiser
S Cordey et al.
Blood virosphere in febrile Tanzanian children.
2021
Emerging microbes & infections
33,929,935
01/05/2021 10:35
01/12/2021 00:00
Cordey
401
10
1
Viral infections are the leading cause of childhood acute febrile illnesses motivating consultation in sub-Saharan Africa. The majority of causal viruses are never identified in low-resource clinical settings as such testing is either not part of routine screening or available diagnostic tools have limited ability to detect new/unexpected viral variants. An in-depth exploration of the blood virome is therefore necessary to clarify the potential viral origin of fever in children. Metagenomic next-generation sequencing is a powerful tool for such broad investigations, allowing the detection of RNA and DNA viral genomes. Here, we describe the blood virome of 816 febrile children (<5 years) presenting at outpatient departments in Dar es Salaam over one-year. We show that half of the patients (394/816) had at least one detected virus recognized as causes of human infection/disease (13.8% enteroviruses (enterovirus A, B, C, and rhinovirus A and C), 12% rotaviruses, 11% human herpesvirus type 6). Additionally, we report the detection of a large number of viruses (related to arthropod, vertebrate or mammalian viral species) not yet known to cause human infection/disease, highlighting those who should be on the radar, deserve specific attention in the febrile paediatric population and, more broadly, for surveillance of emerging pathogens.Trial registration: ClinicalTrials.gov identifier: NCT02225769.
52
P. Baumgaertner, M. Sankar, F. Herrera, F. Benedetti, D. Barras, A.C. Thierry, D. Dangaj, L.E. Kandalaft, G. Coukos, I. Xenarios, N. Guex, A. Harari
P Baumgaertner et al.
Unsupervised Analysis of Flow Cytometry Data in a Clinical Setting Captures Cell Diversity and Allows Population Discovery.
2021
Frontiers in immunology
33,995,353
01/06/2021 18:11
01/01/2021 00:00
Baumgaertner
402
12
Data obtained with cytometry are increasingly complex and their interrogation impacts the type and quality of knowledge gained. Conventional supervised analyses are limited to pre-defined cell populations and do not exploit the full potential of data. Here, in the context of a clinical trial of cancer patients treated with radiotherapy, we performed longitudinal flow cytometry analyses to identify multiple distinct cell populations in circulating whole blood. We cross-compared the results from state-of-the-art recommended supervised analyses with results from MegaClust, a high-performance data-driven clustering algorithm allowing fast and robust identification of cell-type populations. Ten distinct cell populations were accurately identified by supervised analyses, including main T, B, dendritic cell (DC), natural killer (NK) and monocytes subsets. While all ten subsets were also identified with MegaClust, additional cell populations were revealed (e.g. CD4+HLA-DR+ and NKT-like subsets), and DC profiling was enriched by the assignment of additional subset-specific markers. Comparison between transcriptomic profiles of purified DC populations and publicly available datasets confirmed the accuracy of the unsupervised clustering algorithm and demonstrated its potential to identify rare and scarcely described cell subsets. Our observations show that data-driven analyses of cytometry data significantly enrich the amount and quality of knowledge gained, representing an important step in refining the characterization of immune responses.
53
N. Voisin, R.E. Schnur, S. Douzgou, S.M. Hiatt, C.F. Rustad, N.J. Brown, D.L. Earl, B. Keren, O. Levchenko, S. Geuer, S. Verheyen, D. Johnson, Y.A. Zarate, M. Hančárová, D.J. Amor, E.M. Bebin, J. Blatterer, A. Brusco, G. Cappuccio, J. Charrow, N. Chatron, G.M. Cooper, T. Courtin, E. Dadali, J. Delafontaine, E. Del Giudice, M. Doco, G. Douglas, A. Eisenkölbl, T. Funari, G. Giannuzzi, U. Gruber-Sedlmayr, N. Guex, D. Heron, &.#.x.d.8.;.L. Holla, A.C.E. Hurst, J. Juusola, D. Kronn, A. Lavrov, C. Lee, S. Lorrain, E. Merckoll, A. Mikhaleva, J. Norman, S. Pradervand, D. Prchalová, L. Rhodes, V.R. Sanders, Z. Sedláček, H.A. Seebacher, E.A. Sellars, F. Sirchia, T. Takenouchi, A.J. Tanaka, H. Taska-Tench, E. Tønne, K. Tveten, G. Vitiello, M. Vlčková, T. Uehara, C. Nava, B. Yalcin, K. Kosaki, D. Donnai, S. Mundlos, N. Brunetti-Pierri, W.K. Chung, A. Reymond
N Voisin et al.
Variants in the degron of AFF3 are associated with intellectual disability, mesomelic dysplasia, horseshoe kidney, and epileptic encephalopathy.
2021
American journal of human genetics
33,961,779
01/06/2021 18:11
06/05/2021 00:00
Voisin
399
108
5
The ALF transcription factor paralogs, AFF1, AFF2, AFF3, and AFF4, are components of the transcriptional super elongation complex that regulates expression of genes involved in neurogenesis and development. We describe an autosomal dominant disorder associated with de novo missense variants in the degron of AFF3, a nine amino acid sequence important for its binding to ubiquitin ligase, or with de novo deletions of this region. The sixteen affected individuals we identified, along with two previously reported individuals, present with a recognizable pattern of anomalies, which we named KINSSHIP syndrome (KI for horseshoe kidney, NS for Nievergelt/Savarirayan type of mesomelic dysplasia, S for seizures, H for hypertrichosis, I for intellectual disability, and P for pulmonary involvement), partially overlapping the AFF4-associated CHOPS syndrome. Whereas homozygous Aff3 knockout mice display skeletal anomalies, kidney defects, brain malformations, and neurological anomalies, knockin animals modeling one of the microdeletions and the most common of the missense variants identified in affected individuals presented with lower mesomelic limb deformities like KINSSHIP-affected individuals and early lethality, respectively. Overexpression of AFF3 in zebrafish resulted in body axis anomalies, providing some support for the pathological effect of increased amount of AFF3. The only partial phenotypic overlap of AFF3- and AFF4-associated syndromes and the previously published transcriptome analyses of ALF transcription factors suggest that these factors are not redundant and each contributes uniquely to proper development.
54
S. Bassani, E. van Beelen, M. Rossel, N. Voisin, A. Morgan, Y. Arribat, N. Chatron, J. Chrast, M. Cocca, B. Delprat, F. Faletra, G. Giannuzzi, N. Guex, R. Machavoine, S. Pradervand, J.J. Smits, J.M. van de Kamp, A. Ziegler, F. Amati, S. Marlin, H. Kremer, H. Locher, T. Maurice, P. Gasparini, G. Girotto, A. Reymond
S Bassani et al.
Variants in USP48 encoding ubiquitin hydrolase are associated with autosomal dominant non-syndromic hereditary hearing loss.
2021
Human molecular genetics
34,059,922
02/06/2021 10:35
15/09/2021 00:00
Bassani
403
30
19
Non-Syndromic Hereditary Hearing Loss (NSHHL) is a genetically heterogeneous sensory disorder with about 120 genes already associated. Through exome sequencing (ES) and data aggregation, we identified a family with six affected individuals and one unrelated NSHHL patient with predicted-to-be deleterious missense variants in USP48. We also uncovered an eighth patient presenting unilateral cochlear nerve aplasia and a de novo splice variant in the same gene. USP48 encodes a ubiquitin carboxyl-terminal hydrolase under evolutionary constraint. Pathogenicity of the variants is supported by in vitro assays that showed that the mutated proteins are unable to hydrolyze tetra-ubiquitin. Correspondingly, three-dimensional representation of the protein containing the familial missense variant is situated in a loop that might influence the binding to ubiquitin. Consistent with a contribution of USP48 to auditory function, immunohistology showed that the encoded protein is expressed in the developing human inner ear, specifically in the spiral ganglion neurons, outer sulcus, interdental cells of the spiral limbus, stria vascularis, Reissner's membrane and in the transient Kolliker's organ that is essential for auditory development. Engineered zebrafish knocked-down for usp48, the USP48 ortholog, presented with a delayed development of primary motor neurons, less developed statoacoustic neurons innervating the ears, decreased swimming velocity and circling swimming behavior indicative of vestibular dysfunction and hearing impairment. Corroboratingly, acoustic startle response assays revealed a significant decrease of auditory response of zebrafish lacking usp48 at 600 and 800 Hz wavelengths. In conclusion, we describe a novel autosomal dominant NSHHL gene through a multipronged approach combining ES, animal modeling, immunohistology and molecular assays.
55
S. Bibert, N. Guex, J. Lourenco, T. Brahier, M. Papadimitriou-Olivgeris, L. Damonti, O. Manuel, R. Liechti, L. Götz, J. Tschopp, M. Quinodoz, P. Vollenweider, J.L. Pagani, M. Oddo, O. Hügli, F. Lamoth, V. Erard, C. Voide, M. Delorenzi, N. Rufer, F. Candotti, C. Rivolta, N. Boillat-Blanco, P.Y. Bochud,
S Bibert et al.
Transcriptomic Signature Differences Between SARS-CoV-2 and Influenza Virus Infected Patients.
2021
Frontiers in immunology
34,135,895
18/06/2021 10:35
01/01/2021 00:00
Bibert
402
12
The reason why most individuals with COVID-19 have relatively limited symptoms while other develop respiratory distress with life-threatening complications remains unknown. Increasing evidence suggests that COVID-19 associated adverse outcomes mainly rely on dysregulated immunity. Here, we compared transcriptomic profiles of blood cells from 103 patients with different severity levels of COVID-19 with that of 27 healthy and 22 influenza-infected individuals. Data provided a complete overview of SARS-CoV-2-induced immune signature, including a dramatic defect in IFN responses, a reduction of toxicity-related molecules in NK cells, an increased degranulation of neutrophils, a dysregulation of T cells, a dramatic increase in B cell function and immunoglobulin production, as well as an important over-expression of genes involved in metabolism and cell cycle in patients infected with SARS-CoV-2 compared to those infected with influenza viruses. These features also differed according to COVID-19 severity. Overall and specific gene expression patterns across groups can be visualized on an interactive website (https://bix.unil.ch/covid/). Collectively, these transcriptomic host responses to SARS-CoV-2 infection are discussed in the context of current studies, thereby improving our understanding of COVID-19 pathogenesis and shaping the severity level of COVID-19.
56
M. Bruand, D. Barras, M. Mina, E. Ghisoni, M. Morotti, E. Lanitis, N. Fahr, M. Desbuisson, A. Grimm, H. Zhang, C. Chong, J. Dagher, S. Chee, T. Tsianou, J. Dorier, B.J. Stevenson, C. Iseli, C. Ronet, S. Bobisse, R. Genolet, J. Walton, M. Bassani-Sternberg, L.E. Kandalaft, B. Ren, I. McNeish, E. Swisher, A. Harari, M. Delorenzi, G. Ciriello, M. Irving, S. Rusakiewicz, P.G. Foukas, F. Martinon, D. Dangaj Laniti, G. Coukos
M Bruand et al.
Cell-autonomous inflammation of BRCA1-deficient ovarian cancers drives both tumor-intrinsic immunoreactivity and immune resistance via STING.
2021
Cell reports
34,289,354
22/07/2021 10:35
20/07/2021 00:00
Bruand
5
36
3
In this study, we investigate mechanisms leading to inflammation and immunoreactivity in ovarian tumors with homologous recombination deficiency (HRD). BRCA1 loss is found to lead to transcriptional reprogramming in tumor cells and cell-intrinsic inflammation involving type I interferon (IFN) and stimulator of IFN genes (STING). BRCA1-mutated (BRCA1mut) tumors are thus T cell inflamed at baseline. Genetic deletion or methylation of DNA-sensing/IFN genes or CCL5 chemokine is identified as a potential mechanism to attenuate T cell inflammation. Alternatively, in BRCA1mut cancers retaining inflammation, STING upregulates VEGF-A, mediating immune resistance and tumor progression. Tumor-intrinsic STING elimination reduces neoangiogenesis, increases CD8+ T cell infiltration, and reverts therapeutic resistance to dual immune checkpoint blockade (ICB). VEGF-A blockade phenocopies genetic STING loss and synergizes with ICB and/or poly(ADP-ribose) polymerase (PARP) inhibitors to control the outgrowth of Trp53-/-Brca1-/- but not Brca1+/+ ovarian tumors in vivo, offering rational combinatorial therapies for HRD cancers.
57
F. Mazel, L. Malard, H. Niculita-Hirzel, E. Yashiro, H.K. Mod, E.A.D. Mitchell, D. Singer, A. Buri, E. Pinto, N. Guex, E. Lara, A. Guisan
F Mazel et al.
Soil protist function varies with elevation in the Swiss Alps.
2022
Environmental microbiology
34,347,350
05/08/2021 10:35
01/04/2022 00:00
Mazel
404
24
4
Protists are abundant and play key trophic functions in soil. Documenting how their trophic contributions vary across large environmental gradients is essential to understand and predict how biogeochemical cycles will be impacted by global changes. Here, using amplicon sequencing of environmental DNA in open habitat soil from 161 locations spanning 2600 m of elevation in the Swiss Alps (from 400 to 3000 m), we found that, over the whole study area, soils are dominated by consumers, followed by parasites and phototrophs. In contrast, the proportion of these groups in local communities shows large variations in relation to elevation. While there is, on average, three times more consumers than parasites at low elevation (400-1000 m), this ratio increases to 12 at high elevation (2000-3000 m). This suggests that the decrease in protist host biomass and diversity toward mountains tops impact protist functional composition. Furthermore, the taxonomic composition of protists that infect animals was related to elevation while that of protists that infect plants or of protist consumers was related to soil pH. This study provides a first step to document and understand how soil protist functions vary along the elevational gradient.
58
M. Graeff, S. Rana, J.R. Wendrich, J. Dorier, T. Eekhout, A.C. Aliaga Fandino, N. Guex, G.W. Bassel, B. De Rybel, C.S. Hardtke
M Graeff et al.
A single-cell morpho-transcriptomic map of brassinosteroid action in the Arabidopsis root.
2021
Molecular plant
34,358,681
08/08/2021 10:35
06/12/2021 00:00
Graeff
405
14
12
The effects of brassinosteroid signaling on shoot and root development have been characterized in great detail but a simple consistent positive or negative impact on a basic cellular parameter was not identified. In this study, we combined digital 3D single-cell shape analysis and single-cell mRNA sequencing to characterize root meristems and mature root segments of brassinosteroid-blind mutants and wild type. The resultant datasets demonstrate that brassinosteroid signaling affects neither cell volume nor cell proliferation capacity. Instead, brassinosteroid signaling is essential for the precise orientation of cell division planes and the extent and timing of anisotropic cell expansion. Moreover, we found that the cell-aligning effects of brassinosteroid signaling can propagate to normalize the anatomy of both adjacent and distant brassinosteroid-blind cells through non-cell-autonomous functions, which are sufficient to restore growth vigor. Finally, single-cell transcriptome data discern directly brassinosteroid-responsive genes from genes that can react non-cell-autonomously and highlight arabinogalactans as sentinels of brassinosteroid-dependent anisotropic cell expansion.
60
M. Andreatta, F.P.A. David, C. Iseli, N. Guex, S.J. Carmona
M Andreatta et al.
SPICA: Swiss portal for immune cell analysis.
2022
Nucleic acids research
34,747,477
09/11/2021 11:35
07/01/2022 00:00
Andreatta
4
50
D1
Single-cell transcriptomics allows the study of immune cell heterogeneity at an unprecedented level of resolution. The Swiss portal for immune cell analysis (SPICA) is a web resource dedicated to the exploration and analysis of single-cell RNA-seq data of immune cells. In contrast to other single-cell databases, SPICA hosts curated, cell type-specific reference atlases that describe immune cell states at high resolution, and published single-cell datasets analysed in the context of these atlases. Additionally, users can privately analyse their own data in the context of existing atlases and contribute to the SPICA database. SPICA is available at https://spica.unil.ch.
63
J.C. Shillcock, J. Hastings, N. Riguet, H.A. Lashuel
JC Shillcock et al.
Non-monotonic fibril surface occlusion by GFP tags from coarse-grained molecular simulations.
2022
Computational and structural biotechnology journal
35,070,162
25/01/2022 11:35
01/01/2022 00:00
Shillcock
406
20
The pathological growth of amyloid fibrils in neurons underlies the progression of neurodegenerative diseases including Alzheimer's and Parkinson's disease. Fibrils form when soluble monomers oligomerise in the cytoplasm. Their subsequent growth occurs via nucleated polymerization mechanisms involving the free ends of the fibrils augmented by secondary nucleation of new oligomers at their surface. Amyloid fibrils possess a complex interactome with diffusing cytoplasmic proteins that regulates many aspects of their growth, seeding capacity, biochemical activity and transition to pathological inclusions in diseased brains. Changes to their surface are also expected to modify their interactome, pathogenicity and spreading in the brain. Many assays visualise fibril formation, growth and inclusion formation by decorating monomeric proteins with fluorescent tags such as GFP. Recent studies from our group suggest that tags with sizes comparable to the fibril radius may modify the fibril surface accessibility and thus their PTM pattern, interactome and ability to form inclusions. Using coarse-grained molecular simulations of a single alpha synuclein fibril tagged with GFP we find that thermal fluctuations of the tags create a non-monotonic, size-dependent sieve around the fibril that perturbs its interactome with diffusing species. Our results indicate that experiments using tagged and untagged monomers to study the growth and interactome of fibrils should be compared with caution, and the confounding effects of the tags are more complex than a reduction in surface accessibility. The prevalence of fluorescent tags in amyloid fibril growth experiments suggests this has implications beyond the specific alpha synuclein fibrils we model here.
64
H. Li, J. Janssens, M. De Waegeneer, S.S. Kolluru, K. Davie, V. Gardeux, W. Saelens, F.P.A. David, M. Brbić, K. Spanier, J. Leskovec, C.N. McLaughlin, Q. Xie, R.C. Jones, K. Brueckner, J. Shim, S.G. Tattikota, F. Schnorrer, K. Rust, T.G. Nystul, Z. Carvalho-Santos, C. Ribeiro, S. Pal, S. Mahadevaraju, T.M. Przytycka, A.M. Allen, S.F. Goodwin, C.W. Berry, M.T. Fuller, H. White-Cooper, E.L. Matunis, S. DiNardo, A. Galenza, L.E. O'Brien, J.A.T. Dow, , H. Jasper, B. Oliver, N. Perrimon, B. Deplancke, S.R. Quake, L. Luo, S. Aerts, D. Agarwal, Y. Ahmed-Braimah, M. Arbeitman, M.M. Ariss, J. Augsburger, K. Ayush, C.C. Baker, T. Banisch, K. Birker, R. Bodmer, B. Bolival, S.E. Brantley, J.A. Brill, N.C. Brown, N.A. Buehner, X.T. Cai, R. Cardoso-Figueiredo, F. Casares, A. Chang, T.R. Clandinin, S. Crasta, C. Desplan, A.M. Detweiler, D.B. Dhakan, E. Donà, S. Engert, S. Floc'hlay, N. George, A.J. González-Segarra, A.K. Groves, S. Gumbin, Y. Guo, D.E. Harris, Y. Heifetz, S.L. Holtz, F. Horns, B. Hudry, R.J. Hung, Y.N. Jan, J.S. Jaszczak, G.S.X.E. Jefferis, J. Karkanias, T.L. Karr, N.S. Katheder, J. Kezos, A.A. Kim, S.K. Kim, L. Kockel, N. Konstantinides, T.B. Kornberg, H.M. Krause, A.T. Labott, M. Laturney, R. Lehmann, S. Leinwand, J. Li, J.S.S. Li, K. Li, K. Li, L. Li, T. Li, M. Litovchenko, H.H. Liu, Y. Liu, T.C. Lu, J. Manning, A. Mase, M. Matera-Vatnick, N.R. Matias, C.E. McDonough-Goldstein, A. McGeever, A.D. McLachlan, P. Moreno-Roman, N. Neff, M. Neville, S. Ngo, T. Nielsen, C.E. O'Brien, D. Osumi-Sutherland, M.N. Özel, I. Papatheodorou, M. Petkovic, C. Pilgrim, A.O. Pisco, C. Reisenman, E.N. Sanders, G. Dos Santos, K. Scott, A. Sherlekar, P. Shiu, D. Sims, R.V. Sit, M. Slaidina, H.E. Smith, G. Sterne, Y.H. Su, D. Sutton, M. Tamayo, M. Tan, I. Tastekin, C. Treiber, D. Vacek, G. Vogler, S. Waddell, W. Wang, R.I. Wilson, M.F. Wolfner, Y.E. Wong, A. Xie, J. Xu, S. Yamamoto, J. Yan, Z. Yao, K. Yoda, R. Zhu, R.P. Zinzen
H Li et al.
Fly Cell Atlas: A single-nucleus transcriptomic atlas of the adult fruit fly.
2022
Science (New York, N.Y.)
35,239,393
04/03/2022 11:35
04/03/2022 00:00
Li
17
375
6584
For more than 100 years, the fruit fly Drosophila melanogaster has been one of the most studied model organisms. Here, we present a single-cell atlas of the adult fly, Tabula Drosophilae, that includes 580,000 nuclei from 15 individually dissected sexed tissues as well as the entire head and body, annotated to >250 distinct cell types. We provide an in-depth analysis of cell type-related gene signatures and transcription factor markers, as well as sexual dimorphism, across the whole animal. Analysis of common cell types between tissues, such as blood and muscle cells, reveals rare cell types and tissue-specific subtypes. This atlas provides a valuable resource for the Drosophila community and serves as a reference to study genetic perturbations and disease models at single-cell resolution.
68
A. Kaushal, J. Dorier, B. Wang, G. Mohana, M. Taschner, P. Cousin, P. Waridel, C. Iseli, A. Semenova, S. Restrepo, N. Guex, E.L. Aiden, M.C. Gambetta
A Kaushal et al.
Essential role of Cp190 in physical and regulatory boundary formation.
2022
Science advances
35,559,678
15/05/2022 10:35
13/05/2022 00:00
Kaushal
80
8
19
Boundaries in animal genomes delimit contact domains with enhanced internal contact frequencies and have debated functions in limiting regulatory cross-talk between domains and guiding enhancers to target promoters. Most mammalian boundaries form by stalling of chromosomal loop-extruding cohesin by CTCF, but most Drosophila boundaries form CTCF independently. However, how CTCF-independent boundaries form and function remains largely unexplored. Here, we assess genome folding and developmental gene expression in fly embryos lacking the ubiquitous boundary-associated factor Cp190. We find that sequence-specific DNA binding proteins such as CTCF and Su(Hw) directly interact with and recruit Cp190 to form most promoter-distal boundaries. Cp190 is essential for early development and prevents regulatory cross-talk between specific gene loci that pattern the embryo. Cp190 was, in contrast, dispensable for long-range enhancer-promoter communication at tested loci. Cp190 is thus currently the major player in fly boundary formation and function, revealing that diverse mechanisms evolved to partition genomes into independent regulatory domains.
69
V. Scabia, A. Ayyanan, F. De Martino, A. Agnoletto, L. Battista, C. Laszlo, A. Treboux, K. Zaman, A. Stravodimou, D. Jallut, M. Fiche, P. Bucher, G. Ambrosini, G. Sflomos, C. Brisken
V Scabia et al.
Estrogen receptor positive breast cancers have patient specific hormone sensitivities and rely on progesterone receptor.
2022
Nature communications
35,668,111
08/06/2022 10:35
06/06/2022 00:00
Scabia
60
13
1
Estrogen and progesterone receptor (ER, PR) signaling control breast development and impinge on breast carcinogenesis. ER is an established driver of ER + disease but the role of the PR, itself an ER target gene, is debated. We assess the issue in clinically relevant settings by a genetic approach and inject ER + breast cancer cell lines and patient-derived tumor cells to the milk ducts of immunocompromised mice. Such ER + xenografts were exposed to physiologically relevant levels of 17-β-estradiol (E2) and progesterone (P4). We find that independently both premenopausal E2 and P4 levels increase tumor growth and combined treatment enhances metastatic spread. The proliferative responses are patient-specific with MYC and androgen receptor (AR) signatures determining P4 response. PR is required for tumor growth in patient samples and sufficient to drive tumor growth and metastasis in ER signaling ablated tumor cells. Our findings suggest that endocrine therapy may need to be personalized, and that abrogating PR expression can be a therapeutic option.
70
N. Gobet, M. Jan, P. Franken, I. Xenarios
N Gobet et al.
Towards mouse genetic-specific RNA-sequencing read mapping.
2022
PLoS computational biology
36,155,976
27/09/2022 10:35
01/09/2022 00:00
Gobet
77
18
9
Genetic variations affect behavior and cause disease but understanding how these variants drive complex traits is still an open question. A common approach is to link the genetic variants to intermediate molecular phenotypes such as the transcriptome using RNA-sequencing (RNA-seq). Paradoxically, these variants between the samples are usually ignored at the beginning of RNA-seq analyses of many model organisms. This can skew the transcriptome estimates that are used later for downstream analyses, such as expression quantitative trait locus (eQTL) detection. Here, we assessed the impact of reference-based analysis on the transcriptome and eQTLs in a widely-used mouse genetic population: the BXD panel of recombinant inbred lines. We highlight existing reference bias in the transcriptome data analysis and propose practical solutions which combine available genetic variants, genotypes, and genome reference sequence. The use of custom BXD line references improved downstream analysis compared to classical genome reference. These insights would likely benefit genetic studies with a transcriptomic component and demonstrate that genome references need to be reassessed and improved.
71
A. Strembitska, G. Labouèbe, A. Picard, X.P. Berney, D. Tarussio, M. Jan, B. Thorens
A Strembitska et al.
Lipid biosynthesis enzyme Agpat5 in AgRP-neurons is required for insulin-induced hypoglycemia sensing and glucagon secretion.
2022
Nature communications
36,180,454
02/10/2022 10:35
30/09/2022 00:00
Strembitska
60
13
1
The counterregulatory response to hypoglycemia that restores normal blood glucose levels is an essential physiological function. It is initiated, in large part, by incompletely characterized brain hypoglycemia sensing neurons that trigger the secretion of counterregulatory hormones, in particular glucagon, to stimulate hepatic glucose production. In a genetic screen of recombinant inbred BXD mice we previously identified Agpat5 as a candidate regulator of hypoglycemia-induced glucagon secretion. Here, using genetic mouse models, we demonstrate that Agpat5 expressed in agouti-related peptide neurons is required for their activation by hypoglycemia, for hypoglycemia-induced vagal nerve activity, and glucagon secretion. We find that inactivation of Agpat5 leads to increased fatty acid oxidation and ATP production and that suppressing Cpt1a-dependent fatty acid import into mitochondria restores hypoglycemia sensing. Collectively, our data show that AgRP neurons are involved in the control of glucagon secretion and that Agpat5, by partitioning fatty acyl-CoAs away from mitochondrial fatty acid oxidation and ATP generation, ensures that the fall in intracellular ATP, which triggers neuronal firing, faithfully reflects changes in glycemia.
72
A.P. Machado, A. Topaloudis, T. Cumer, E. Lavanchy, V. Bontzorlos, R. Ceccherelli, M. Charter, N. Kassinis, P. Lymberakis, F. Manzia, A.L. Ducrest, M. Dupasquier, N. Guex, A. Roulin, J. Goudet
AP Machado et al.
Genomic consequences of colonisation, migration and genetic drift in barn owl insular populations of the eastern Mediterranean.
2022
Molecular ecology
34,894,026
20/10/2022 15:49
01/03/2022 00:00
Machado
407
31
5
The study of insular populations was key in the development of evolutionary theory. The successful colonisation of an island depends on the geographic context, and specific characteristics of the organism and the island, but also on stochastic processes. As a result, apparently identical islands may harbour populations with contrasting histories. Here, we use whole genome sequences of 65 barn owls to investigate the patterns of inbreeding and genetic diversity of insular populations in the eastern Mediterranean Sea. We focus on Crete and Cyprus, islands with similar size, climate and distance to mainland, that provide natural replicates for a comparative analysis of the impacts of microevolutionary processes on isolated populations. We show that barn owl populations from each island have a separate origin, Crete being genetically more similar to other Greek islands and mainland Greece, and Cyprus more similar to the Levant. Further, our data show that their respective demographic histories following colonisation were also distinct. On the one hand, Crete harbours a small population and maintains very low levels of gene flow with neighbouring populations. This has resulted in low genetic diversity, strong genetic drift, increased relatedness in the population and remote inbreeding. Cyprus, on the other hand, appears to maintain enough gene flow with the mainland to avoid such an outcome. Our study provides a comparative population genomic analysis of the effects of neutral processes on a classical island-mainland model system. It provides empirical evidence for the role of stochastic processes in determining the fate of diverging isolated populations.
73
N. Riguet, A.L. Mahul-Mellier, N. Maharjan, J. Burtscher, M. Croisier, G. Knott, J. Hastings, A. Patin, V. Reiterer, H. Farhan, S. Nasarov, H.A. Lashuel
N Riguet et al.
Nuclear and cytoplasmic huntingtin inclusions exhibit distinct biochemical composition, interactome and ultrastructural properties.
2021
Nature communications
34,772,920
20/10/2022 15:49
12/11/2021 00:00
Riguet
60
12
1
Despite the strong evidence linking the aggregation of the Huntingtin protein (Htt) to the pathogenesis of Huntington's disease (HD), the mechanisms underlying Htt aggregation and neurodegeneration remain poorly understood. Herein, we investigated the ultrastructural properties and protein composition of Htt cytoplasmic and nuclear inclusions in mammalian cells and primary neurons overexpressing mutant exon1 of the Htt protein. Our findings provide unique insight into the ultrastructural properties of cytoplasmic and nuclear Htt inclusions and their mechanisms of formation. We show that Htt inclusion formation and maturation are complex processes that, although initially driven by polyQ-dependent Htt aggregation, also involve the polyQ and PRD domain-dependent sequestration of lipids and cytoplasmic and cytoskeletal proteins related to HD dysregulated pathways; the recruitment and accumulation of remodeled or dysfunctional membranous organelles, and the impairment of the protein quality control and degradation machinery. We also show that nuclear and cytoplasmic Htt inclusions exhibit distinct biochemical compositions and ultrastructural properties, suggesting different mechanisms of aggregation and toxicity.
74
A.P. Machado, T. Cumer, C. Iseli, E. Beaudoing, A.L. Ducrest, M. Dupasquier, N. Guex, K. Dichmann, R. Lourenço, J. Lusby, H.D. Martens, L. Prévost, D. Ramsden, A. Roulin, J. Goudet
AP Machado et al.
Unexpected post-glacial colonisation route explains the white colour of barn owls (Tyto alba) from the British Isles.
2022
Molecular ecology
34,695,244
20/10/2022 15:49
01/01/2022 00:00
Machado
407
31
2
The climate fluctuations of the Quaternary shaped the movement of species in and out of glacial refugia. In Europe, the majority of species followed one of the described traditional postglacial recolonization routes from the southern peninsulas towards the north. Like most organisms, barn owls are assumed to have colonized the British Isles by crossing over Doggerland, a land bridge that connected Britain to northern Europe. However, while they are dark rufous in northern Europe, barn owls in the British Isles are conspicuously white, a contrast that could suggest selective forces are at play on the islands. Yet, our analysis of known candidate genes involved in coloration found no signature of selection. Instead, using whole genome sequences and species distribution modelling, we found that owls colonised the British Isles soon after the last glaciation, directly from a white coloured refugium in the Iberian Peninsula, before colonising northern Europe. They would have followed a hitherto unknown post-glacial colonization route to the Isles over a westwards path of suitable habitat in now submerged land in the Bay of Biscay, thus not crossing Doggerland. As such, they inherited the white colour of their Iberian founders and maintained it through low gene flow with the mainland that prevents the import of rufous alleles. Thus, we contend that neutral processes probably explain this contrasting white colour compared to continental owls. With the barn owl being a top predator, we expect future research will show this unanticipated route was used by other species from its paleo community.
75
H.A. Lashuel, A.L. Mahul-Mellier, S. Novello, R.N. Hegde, Y. Jasiqi, M.F. Altay, S. Donzelli, S.M. DeGuire, R. Burai, P. Magalhães, A. Chiki, J. Ricci, M. Boussouf, A. Sadek, E. Stoops, C. Iseli, N. Guex
HA Lashuel et al.
Revisiting the specificity and ability of phospho-S129 antibodies to capture alpha-synuclein biochemical and pathological diversity.
2022
NPJ Parkinson's disease
36,266,318
21/10/2022 10:35
20/10/2022 00:00
Lashuel
408
8
1
Antibodies against phosphorylated alpha-synuclein (aSyn) at S129 have emerged as the primary tools to investigate, monitor, and quantify aSyn pathology in the brain and peripheral tissues of patients with Parkinson's disease and other neurodegenerative diseases. Herein, we demonstrate that the co-occurrence of multiple pathology-associated C-terminal post-translational modifications (PTMs) (e.g., phosphorylation at Tyrosine 125 or truncation at residue 133 or 135) differentially influences the detection of pS129-aSyn species by pS129-aSyn antibodies. These observations prompted us to systematically reassess the specificity of the most commonly used pS129 antibodies against monomeric and aggregated forms of pS129-aSyn in mouse brain slices, primary neurons, mammalian cells and seeding models of aSyn pathology formation. We identified two antibodies that are insensitive to pS129 neighboring PTMs. Although most pS129 antibodies showed good performance in detecting aSyn aggregates in cells, neurons and mouse brain tissue containing abundant aSyn pathology, they also showed cross-reactivity towards other proteins and often detected non-specific low and high molecular weight bands in aSyn knock-out samples that could be easily mistaken for monomeric or high molecular weight aSyn species. Our observations suggest that not all pS129 antibodies capture the biochemical and morphological diversity of aSyn pathology, and all should be used with the appropriate protein standards and controls when investigating aSyn under physiological conditions. Finally, our work underscores the need for more pS129 antibodies that are not sensitive to neighboring PTMs and more thorough characterization and validation of existing and new antibodies.
76
P.G. Manti, F. Darbellay, M. Leleu, A.Y. Coughlan, B. Moret, J. Cuennet, F. Droux, M. Stoudmann, G.F. Mancini, A. Hautier, J. Sordet-Dessimoz, S.D. Vincent, G. Testa, G. Cossu, Y. Barrandon
PG Manti et al.
The Transcriptional Regulator Prdm1 Is Essential for the Early Development of the Sensory Whisker Follicle and Is Linked to the Beta-Catenin First Dermal Signal.
2022
Biomedicines
36,289,911
28/10/2022 10:35
20/10/2022 00:00
Manti
409
10
10
Prdm1 mutant mice are one of the rare mutant strains that do not develop whisker hair follicles while still displaying a pelage. Here, we show that Prdm1 is expressed at the earliest stage of whisker development in clusters of mesenchymal cells before placode formation. Its conditional knockout in the murine soma leads to the loss of expression of Bmp2, Shh, Bmp4, Krt17, Edar, and Gli1, though leaving the β-catenin-driven first dermal signal intact. Furthermore, we show that Prdm1 expressing cells not only act as a signaling center but also as a multipotent progenitor population contributing to the several lineages of the adult whisker. We confirm by genetic ablation experiments that the absence of macro vibrissae reverberates on the organization of nerve wiring in the mystacial pads and leads to the reorganization of the barrel cortex. We demonstrate that Lef1 acts upstream of Prdm1 and identify a primate-specific deletion of a Lef1 enhancer named Leaf. This loss may have been significant in the evolutionary process, leading to the progressive defunctionalization and disappearance of vibrissae in primates.
78
A. Strefeler, M. Jan, M. Quadroni, T. Teav, N. Rosenberg, J.Y. Chatton, N. Guex, H. Gallart-Ayala, J. Ivanisevic
A Strefeler et al.
Molecular insights into sex-specific metabolic alterations in Alzheimer's mouse brain using multi-omics approach.
2023
Alzheimer's research & therapy
36,624,525
10/01/2023 11:35
09/01/2023 00:00
Strefeler
410
15
1
Alzheimer's disease (AD) is a progressive neurodegenerative disorder that is characterized by altered cellular metabolism in the brain. Several of these alterations have been found to be exacerbated in females, known to be disproportionately affected by AD. We aimed to unravel metabolic alterations in AD at the metabolic pathway level and evaluate whether they are sex-specific through integrative metabolomic, lipidomic, and proteomic analysis of mouse brain tissue.
80
R. Sarkis, O. Burri, C. Royer-Chardon, F. Schyrr, S. Blum, M. Costanza, S. Cherix, N. Piazzon, C. Barcena, B. Bisig, V. Nardi, R. Sarro, G. Ambrosini, M. Weigert, O. Spertini, S. Blum, B. Deplancke, A. Seitz, L. de Leval, O. Naveiras
R Sarkis et al.
MarrowQuant 2.0: A Digital Pathology Workflow Assisting Bone Marrow Evaluation in Experimental and Clinical Hematology.
2023
Modern pathology : an official journal of the United States and Canadian Academy of Pathology, Inc
36,788,087
15/02/2023 11:35
01/04/2023 00:00
Sarkis
411
36
4
Bone marrow (BM) cellularity assessment is a crucial step in the evaluation of BM trephine biopsies for hematologic and nonhematologic disorders. Clinical assessment is based on a semiquantitative visual estimation of the hematopoietic and adipocytic components by hematopathologists, which does not provide quantitative information on other stromal compartments. In this study, we developed and validated MarrowQuant 2.0, an efficient, user-friendly digital hematopathology workflow integrated within QuPath software, which serves as BM quantifier for 5 mutually exclusive compartments (bone, hematopoietic, adipocytic, and interstitial/microvasculature areas and other) and derives the cellularity of human BM trephine biopsies. Instance segmentation of individual adipocytes is realized through the adaptation of the machine-learning-based algorithm StarDist. We calculated BM compartments and adipocyte size distributions of hematoxylin and eosin images obtained from 250 bone specimens, from control subjects and patients with acute myeloid leukemia or myelodysplastic syndrome, at diagnosis and follow-up, and measured the agreement of cellularity estimates by MarrowQuant 2.0 against visual scores from 4 hematopathologists. The algorithm was capable of robust BM compartment segmentation with an average mask accuracy of 86%, maximal for bone (99%), hematopoietic (92%), and adipocyte (98%) areas. MarrowQuant 2.0 cellularity score and hematopathologist estimations were highly correlated (R2 = 0.92-0.98, intraclass correlation coefficient [ICC] = 0.98; interobserver ICC = 0.96). BM compartment segmentation quantitatively confirmed the reciprocity of the hematopoietic and adipocytic compartments. MarrowQuant 2.0 performance was additionally tested for cellularity assessment of specimens prospectively collected from clinical routine diagnosis. After special consideration for the choice of the cellularity equation in specimens with expanded stroma, performance was similar in this setting (R2 = 0.86, n = 42). Thus, we conclude that these validation experiments establish MarrowQuant 2.0 as a reliable tool for BM cellularity assessment. We expect this workflow will serve as a clinical research tool to explore novel biomarkers related to BM stromal components and may contribute to further validation of future digitalized diagnostic hematopathology workstreams.
81
B. Conrad, C. Iseli, M. Pirovino
B Conrad et al.
Energy-harnessing problem solving of primordial life: Modeling the emergence of catalytic host-nested parasite life cycles.
2023
PloS one
36,972,235
28/03/2023 10:35
01/01/2023 00:00
Conrad
6
18
3
All life forms on earth ultimately descended from a primordial population dubbed the last universal common ancestor or LUCA via Darwinian evolution. Extant living systems share two salient functional features, a metabolism extracting and transforming energy required for survival, and an evolvable, informational polymer-the genome-conferring heredity. Genome replication invariably generates essential and ubiquitous genetic parasites. Here we model the energetic, replicative conditions of LUCA-like organisms and their parasites, as well as adaptive problem solving of host-parasite pairs. We show using an adapted Lotka-Volterra frame-work that three host-parasite pairs-individually a unit of a host and a parasite that is itself parasitized, therefore a nested parasite pair-are sufficient for robust and stable homeostasis, forming a life cycle. This nested parasitism model includes competition and habitat restriction. Its catalytic life cycle efficiently captures, channels and transforms energy, enabling dynamic host survival and adaptation. We propose a Malthusian fitness model for a quasispecies evolving through a host-nested parasite life cycle with two core features, rapid replacement of degenerate parasites and increasing evolutionary stability of host-nested parasite units from one to three pairs.
82
N. Ibrahim, S. Naz, F. Mattioli, N. Guex, S. Sharif, A. Iqbal, M. Ansar, A. Reymond
N Ibrahim et al.
A Biallelic Truncating Variant in the TPR Domain of GEMIN5 Associated with Intellectual Disability and Cerebral Atrophy.
2023
Genes
36,980,979
30/03/2023 10:35
13/03/2023 00:00
Ibrahim
412
14
3
GEMIN5 is a multifunctional RNA-binding protein required for the assembly of survival motor neurons. Several bi-allelic truncating and missense variants in this gene are reported to cause a neurodevelopmental disorder characterized by cerebellar atrophy, intellectual disability (ID), and motor dysfunction. Whole exome sequencing of a Pakistani consanguineous family with three brothers affected by ID, cerebral atrophy, mobility, and speech impairment revealed a novel homozygous 3bp-deletion NM_015465.5:c.3162_3164del that leads to the loss of NM_015465.5 (NP_056280.2):p. (Asp1054_Ala1055delinsGlu) amino acid in one of the α-helixes of the tetratricopeptide repeats of GEMIN5. In silico 3D representations of the GEMIN5 dimerization domain show that this variant likely affects the orientation of the downstream sidechains out of the helix axis, which would affect the packing with neighboring helices. The phenotype of all affected siblings overlaps well with previously reported patients, suggesting that NM_015465.5: c.3162_3164del (NP_056280.2):p. (Asp1054_Ala1055delinsGlu) is a novel GEMIN5 pathogenic variant. Overall, our data expands the molecular and clinical phenotype of the recently described neurodevelopmental disorder with cerebellar atrophy and motor dysfunction (NEDCAM) syndrome.
83
N. Vesel, C. Iseli, N. Guex, A. Lemopoulos, M. Blokesch
N Vesel et al.
DNA modifications impact natural transformation of Acinetobacter baumannii.
2023
Nucleic acids research
37,178,001
14/05/2023 10:35
23/06/2023 00:00
Vesel
4
51
11
Acinetobacter baumannii is a dangerous nosocomial pathogen, especially due to its ability to rapidly acquire new genetic traits, including antibiotic resistance genes (ARG). In A. baumannii, natural competence for transformation, one of the primary modes of horizontal gene transfer (HGT), is thought to contribute to ARG acquisition and has therefore been intensively studied. However, knowledge regarding the potential role of epigenetic DNA modification(s) on this process remains lacking. Here, we demonstrate that the methylome pattern of diverse A. baumannii strains differs substantially and that these epigenetic marks influence the fate of transforming DNA. Specifically, we describe a methylome-dependent phenomenon that impacts intra- and inter-species DNA exchange by the competent A. baumannii strain A118. We go on to identify and characterize an A118-specific restriction-modification (RM) system that impairs transformation when the incoming DNA lacks a specific methylation signature. Collectively, our work contributes towards a more holistic understanding of HGT in this organism and may also aid future endeavors towards tackling the spread of novel ARGs. In particular, our results suggest that DNA exchanges between bacteria that share similar epigenomes are favored and could therefore guide future research into identifying the reservoir(s) of dangerous genetic traits for this multi-drug resistant pathogen.
84
F. Mattioli, L. Worpenberg, C.T. Li, N. Ibrahim, S. Naz, S. Sharif, S.G. Firouzabadi, S. Vosoogh, R. Saraeva-Lamri, L. Raymond, C. Trujillo, N. Guex, S.E. Antonarakis, M. Ansar, H. Darvish, R.J. Liu, J.Y. Roignant, A. Reymond
F Mattioli et al.
Biallelic variants in NSUN6 cause an autosomal recessive neurodevelopmental disorder.
2023
Genetics in medicine : official journal of the American College of Medical Genetics
37,226,891
26/05/2023 10:35
01/09/2023 00:00
Mattioli
413
25
9
5-methylcytosine RNA modifications are driven by NSUN methyltransferases. Although variants in NSUN2 and NSUN3 were associated with neurodevelopmental diseases, the physiological role of NSUN6 modifications on transfer RNAs and messenger RNAs remained elusive.
87
G. Mohana, J. Dorier, X. Li, M. Mouginot, R.C. Smith, H. Malek, M. Leleu, D. Rodriguez, J. Khadka, P. Rosa, P. Cousin, C. Iseli, S. Restrepo, N. Guex, B.D. McCabe, A. Jankowski, M.S. Levine, M.C. Gambetta
G Mohana et al.
Chromosome-level organization of the regulatory genome in the Drosophila nervous system.
2023
Cell
37,536,338
04/08/2023 10:35
31/08/2023 00:00
Mohana
112
186
18
Previous studies have identified topologically associating domains (TADs) as basic units of genome organization. We present evidence of a previously unreported level of genome folding, where distant TAD pairs, megabases apart, interact to form meta-domains. Within meta-domains, gene promoters and structural intergenic elements present in distant TADs are specifically paired. The associated genes encode neuronal determinants, including those engaged in axonal guidance and adhesion. These long-range associations occur in a large fraction of neurons but support transcription in only a subset of neurons. Meta-domains are formed by diverse transcription factors that are able to pair over long and flexible distances. We present evidence that two such factors, GAF and CTCF, play direct roles in this process. The relative simplicity of higher-order meta-domain interactions in Drosophila, compared with those previously described in mammals, allowed the demonstration that genomes can fold into highly specialized cell-type-specific scaffolds that enable megabase-scale regulatory associations.
89
S. Bobisse, V. Bianchi, J.L. Tanyi, A. Sarivalasis, E. Missiaglia, R. Pétremand, F. Benedetti, D.A. Torigian, R. Genolet, D. Barras, A. Michel, S.A. Mastroyannis, E. Zsiros, D. Dangaj Laniti, Z. Tsourti, B.J. Stevenson, C. Iseli, B.L. Levine, D.E. Speiser, D. Gfeller, M. Bassani-Sternberg, D.J. Powell, C.H. June, U. Dafni, L.E. Kandalaft, A. Harari, G. Coukos
S Bobisse et al.
A phase 1 trial of adoptive transfer of vaccine-primed autologous circulating T cells in ovarian cancer.
2023
Nature cancer
37,735,588
23/09/2023 10:35
01/10/2023 00:00
Bobisse
414
4
10
We have previously shown that vaccination with tumor-pulsed dendritic cells amplifies neoantigen recognition in ovarian cancer. Here, in a phase 1 clinical study ( NCT01312376 /UPCC26810) including 19 patients, we show that such responses are further reinvigorated by subsequent adoptive transfer of vaccine-primed, ex vivo-expanded autologous peripheral blood T cells. The treatment is safe, and epitope spreading with novel neopeptide reactivities was observed after cell infusion in patients who experienced clinical benefit, suggesting reinvigoration of tumor-sculpting immunity.
90
C. Nunes, S. Proença, G. Ambrosini, D. Pamies, A. Thomas, N.I. Kramer, M.G. Zurich
C Nunes et al.
Integrating distribution kinetics and toxicodynamics to assess repeat dose neurotoxicity in vitro using human BrainSpheres: a case study on amiodarone.
2023
Frontiers in pharmacology
37,745,076
26/09/2023 10:35
01/01/2023 00:00
Nunes
415
14
For ethical, economical, and scientific reasons, animal experimentation, used to evaluate the potential neurotoxicity of chemicals before their release in the market, needs to be replaced by new approach methodologies. To illustrate the use of new approach methodologies, the human induced pluripotent stem cell-derived 3D model BrainSpheres was acutely (48 h) or repeatedly (7 days) exposed to amiodarone (0.625-15 µM), a lipophilic antiarrhythmic drug reported to have deleterious effects on the nervous system. Neurotoxicity was assessed using transcriptomics, the immunohistochemistry of cell type-specific markers, and real-time reverse transcription-polymerase chain reaction for various genes involved in the lipid metabolism. By integrating distribution kinetics modeling with neurotoxicity readouts, we show that the observed time- and concentration-dependent increase in the neurotoxic effects of amiodarone is driven by the cellular accumulation of amiodarone after repeated dosing. The development of a compartmental in vitro distribution kinetics model allowed us to predict the change in cell-associated concentrations in BrainSpheres with time and for different exposure scenarios. The results suggest that human cells are intrinsically more sensitive to amiodarone than rodent cells. Amiodarone-induced regulation of lipid metabolism genes was observed in brain cells for the first time. Astrocytes appeared to be the most sensitive human brain cell type in vitro. In conclusion, assessing readouts at different molecular levels after the repeat dosing of human induced pluripotent stem cell-derived BrainSpheres in combination with the compartmental modeling of in vitro kinetics provides a mechanistic means to assess neurotoxicity pathways and refine chemical safety assessment for humans.
91
R. Mishra, M. Hannebelle, V.P. Patil, A. Dubois, C. Garcia-Mouton, G.M. Kirsch, M. Jan, K. Sharma, N. Guex, J. Sordet-Dessimoz, J. Perez-Gil, M. Prakash, G.W. Knott, N. Dhar, J.D. McKinney, V.V. Thacker
R Mishra et al.
Mechanopathology of biofilm-like Mycobacterium tuberculosis cords.
2023
Cell
37,865,090
22/10/2023 10:36
09/11/2023 00:00
Mishra
112
186
23
Mycobacterium tuberculosis (Mtb) cultured axenically without detergent forms biofilm-like cords, a clinical identifier of virulence. In lung-on-chip (LoC) and mouse models, cords in alveolar cells contribute to suppression of innate immune signaling via nuclear compression. Thereafter, extracellular cords cause contact-dependent phagocyte death but grow intercellularly between epithelial cells. The absence of these mechanopathological mechanisms explains the greater proportion of alveolar lesions with increased immune infiltration and dissemination defects in cording-deficient Mtb infections. Compression of Mtb lipid monolayers induces a phase transition that enables mechanical energy storage. Agent-based simulations demonstrate that the increased energy storage capacity is sufficient for the formation of cords that maintain structural integrity despite mechanical perturbation. Bacteria in cords remain translationally active despite antibiotic exposure and regrow rapidly upon cessation of treatment. This study provides a conceptual framework for the biophysics and function in tuberculosis infection and therapy of cord architectures independent of mechanisms ascribed to single bacteria.
92
M. Pirovino, C. Iseli, J.A. Curran, B. Conrad
M Pirovino et al.
Biomathematical enzyme kinetics model of prebiotic autocatalytic RNA networks: degenerating parasite-specific hyperparasite catalysts confer parasite resistance and herald the birth of molecular immunity.
2025
PLoS computational biology
39,752,624
22/01/2025 11:12
01/01/2025 00:00
Pirovino
77
21
1
Catalysis and specifically autocatalysis are the quintessential building blocks of life. Yet, although autocatalytic networks are necessary, they are not sufficient for the emergence of life-like properties, such as replication and adaptation. The ultimate and potentially fatal threat faced by molecular replicators is parasitism; if the polymerase error rate exceeds a critical threshold, even the fittest molecular species will disappear. Here we have developed an autocatalytic RNA early life mathematical network model based on enzyme kinetics, specifically the steady-state approximation. We confirm previous models showing that these second-order autocatalytic cycles are sustainable, provided there is a sufficient nucleotide pool. However, molecular parasites become untenable unless they sequentially degenerate to hyperparasites (i.e. parasites of parasites). Parasite resistance-a parasite-specific host response decreasing parasite fitness-is acquired gradually, and eventually involves an increased binding affinity of hyperparasites for parasites. Our model is supported at three levels; firstly, ribozyme polymerases display Michaelis-Menten saturation kinetics and comply with the steady-state approximation. Secondly, ribozyme polymerases are capable of sustainable auto-amplification and of surmounting the fatal error threshold. Thirdly, with growing sequence divergence of host and parasite catalysts, the probability of self-binding is expected to increase and the trend towards cross-reactivity to diminish. Our model predicts that primordial host-RNA populations evolved via an arms race towards a host-parasite-hyperparasite catalyst trio that conferred parasite resistance within an RNA replicator niche. While molecular parasites have traditionally been viewed as a nuisance, our model argues for their integration into the host habitat rather than their separation. It adds another mechanism-with biochemical precision-by which parasitism can be tamed and offers an attractive explanation for the universal coexistence of catalyst trios within prokaryotes and the virosphere, heralding the birth of a primitive molecular immunity.
93
R. Meurs, M. De Matos, A. Bothe, N. Guex, T. Weber, A.A. Teleman, N. Ban, D. Gatfield
R Meurs et al.
MCTS2 and distinct eIF2D roles in uORF-dependent translation regulation revealed by in vitro re-initiation assays.
2025
The EMBO journal
39,748,120
22/01/2025 11:12
01/02/2025 00:00
Meurs
9
44
3
Ribosomes scanning from the mRNA 5' cap to the start codon may initiate at upstream open reading frames (uORFs), decreasing protein biosynthesis. Termination at a uORF can lead to re-initiation, where 40S subunits resume scanning and initiate another translation event downstream. The noncanonical translation factors MCTS1-DENR participate in re-initiation at specific uORFs, but knowledge of other trans-acting factors or uORF features influencing re-initiation is limited. Here, we establish a cell-free re-initiation assay using HeLa lysates to address this question. Comparing in vivo and in vitro re-initiation on uORF-containing reporters, we validate MCTS1-DENR-dependent re-initiation in vitro. Using this system and ribosome profiling in cells, we found that knockdown of the MCTS1-DENR homolog eIF2D causes widespread gene deregulation unrelated to uORF translation, and thus distinct to MCTS1-DENR-dependent re-initiation regulation. Additionally, we identified MCTS2, encoded by an Mcts1 retrogene, as a DENR partner promoting re-initiation in vitro, providing a plausible explanation for clinical differences associated with DENR vs. MCTS1 mutations in humans.
94
A. Tonelli, P. Cousin, A. Jankowski, B. Wang, J. Dorier, J. Barraud, S. Zunjarrao, M.C. Gambetta
A Tonelli et al.
Systematic screening of enhancer-blocking insulators in Drosophila identifies their DNA sequence determinants.
2025
Developmental cell
39,532,105
22/01/2025 11:12
24/02/2025 00:00
Tonelli
19
60
4
Long-range transcriptional activation of gene promoters by abundant enhancers in animal genomes calls for mechanisms to limit inappropriate regulation. DNA elements called insulators serve this purpose by shielding promoters from an enhancer when interposed. Unlike promoters and enhancers, insulators have not been systematically characterized due to lacking high-throughput screening assays, and questions regarding how insulators are distributed and encoded in the genome remain. Here, we establish "insulator-seq" as a plasmid-based massively parallel reporter assay in Drosophila cultured cells to perform a systematic insulator screen of selected genomic loci. Screening developmental gene loci showed that not all insulator protein binding sites effectively block enhancer-promoter communication. Deep insulator mutagenesis identified sequences flexibly positioned around the CTCF insulator protein binding motif that are critical for functionality. The ability to screen millions of DNA sequences without positional effect has enabled functional mapping of insulators and provided further insights into the determinants of insulators.
95
V. Gardeux, R.P.J. Bevers, F.P.A. David, E. Rosschaert, R. Rochepeau, B. Deplancke
V Gardeux et al.
DGRPool, a web tool leveraging harmonized Drosophila Genetic Reference Panel phenotyping data for the study of complex traits.
2024
eLife
39,431,984
22/01/2025 11:12
21/10/2024 00:00
Gardeux
15
12
Genome-wide association studies have advanced our understanding of complex traits, but studying how a GWAS variant can affect a specific trait in the human population remains challenging due to environmental variability. Drosophila melanogaster is in this regard an excellent model organism for studying the relationship between genetic and phenotypic variation due to its simple handling, standardized growth conditions, low cost, and short lifespan. The Drosophila Genetic Reference Panel (DGRP) in particular has been a valuable tool for studying complex traits, but proper harmonization and indexing of DGRP phenotyping data is necessary to fully capitalize on this resource. To address this, we created a web tool called DGRPool (dgrpool.epfl.ch), which aggregates phenotyping data of 1034 phenotypes across 135 DGRP studies in a common environment. DGRPool enables users to download data and run various tools such as genome-wide (GWAS) and phenome-wide (PheWAS) association studies. As a proof-of-concept, DGRPool was used to study the longevity phenotype and uncovered both established and unexpected correlations with other phenotypes such as locomotor activity, starvation resistance, desiccation survival, and oxidative stress resistance. DGRPool has the potential to facilitate new genetic and molecular insights of complex traits in Drosophila and serve as a valuable, interactive tool for the scientific community.
96
S. Martin, F. Fournes, G. Ambrosini, C. Iseli, K. Bojkowska, J. Marquis, N. Guex, J. Collier
S Martin et al.
DNA methylation by CcrM contributes to genome maintenance in the Agrobacterium tumefaciens plant pathogen.
2024
Nucleic acids research
39,228,370
22/01/2025 11:12
28/10/2024 00:00
Martin
4
52
19
The cell cycle-regulated DNA methyltransferase CcrM is conserved in most Alphaproteobacteria, but its role in bacteria with complex or multicentric genomes remains unexplored. Here, we compare the methylome, the transcriptome and the phenotypes of wild-type and CcrM-depleted Agrobacterium tumefaciens cells with a dicentric chromosome with two essential replication origins. We find that DNA methylation has a pleiotropic impact on motility, biofilm formation and viability. Remarkably, CcrM promotes the expression of the repABCCh2 operon, encoding proteins required for replication initiation/partitioning at ori2, and represses gcrA, encoding a conserved global cell cycle regulator. Imaging ori1 and ori2 in live cells, we show that replication from ori2 is often delayed in cells with a hypo-methylated genome, while ori2 over-initiates in cells with a hyper-methylated genome. Further analyses show that GcrA promotes the expression of the RepCCh2 initiator, most likely through the repression of a RepECh2 anti-sense RNA. Altogether, we propose that replication at ori1 leads to a transient hemi-methylation and activation of the gcrA promoter, allowing repCCh2 activation by GcrA and contributing to initiation at ori2. This study then uncovers a novel and original connection between CcrM-dependent DNA methylation, a conserved epigenetic regulator and genome maintenance in an Alphaproteobacterial pathogen.
97
G. Santoni, S. Astori, M. Leleu, L. Glauser, S.A. Zamora, M. Schioppa, I. Tarulli, C. Sandi, J. Gräff
G Santoni et al.
Chromatin plasticity predetermines neuronal eligibility for memory trace formation.
2024
Science (New York, N.Y.)
39,052,786
22/01/2025 11:12
26/07/2024 00:00
Santoni
17
385
6707
Memories are encoded by sparse populations of neurons but how such sparsity arises remains largely unknown. We found that a neuron's eligibility to be recruited into the memory trace depends on its epigenetic state prior to encoding. Principal neurons in the mouse lateral amygdala display intrinsic chromatin plasticity, which when experimentally elevated favors neuronal allocation into the encoding ensemble. Such chromatin plasticity occurred at genomic regions underlying synaptic plasticity and was accompanied by increased neuronal excitability in single neurons in real time. Lastly, optogenetic silencing of the epigenetically altered neurons prevented memory expression, revealing a cell-autonomous relationship between chromatin plasticity and memory trace formation. These results identify the epigenetic state of a neuron as a key factor enabling information encoding.
98
C. Lorin, R. Guiet, N. Chiaruttini, G. Ambrosini, E. Boci, M. Abdellah, H. Markram, D. Keller
C Lorin et al.
Structural and molecular characterization of astrocyte and vasculature connectivity in the mouse hippocampus and cortex.
2024
Glia
39,007,459
22/01/2025 11:12
01/11/2024 00:00
Lorin
416
72
11
The relation of astrocytic endfeet to the vasculature plays a key functional role in the neuro-glia-vasculature unit. We characterize the spatial organization of astrocytes and the structural aspects that facilitate their involvement in molecular exchanges. Using double transgenic mice, we performed co-immunostaining, confocal microscopy, and three-dimensional digital segmentation to investigate the biophysical and molecular organization of astrocytes and their intricate endfoot network at the micrometer level in the isocortex and hippocampus. The results showed that hippocampal astrocytes had smaller territories, reduced endfoot dimensions, and fewer contacts with blood vessels compared with those in the isocortex. Additionally, we found that both connexins 43 and 30 have a higher density in the endfoot and the former is overexpressed relative to the latter. However, due to the limitations of the method, further studies are needed to determine the exact localization on the endfoot. The quantitative information obtained in this study will be useful for modeling the interactions of astrocytes with the vasculature.
99
M. Jan, S. Jimenez, C.N. Hor, D.J. Dijk, A.C. Skeldon, P. Franken
M Jan et al.
Model integration of circadian- and sleep-wake-driven contributions to rhythmic gene expression reveals distinct regulatory principles.
2024
Cell systems
38,986,625
22/01/2025 11:12
17/07/2024 00:00
Jan
417
15
7
Analyses of gene-expression dynamics in research on circadian rhythms and sleep homeostasis often describe these two processes using separate models. Rhythmically expressed genes are, however, likely to be influenced by both processes. We implemented a driven, damped harmonic oscillator model to estimate the contribution of circadian- and sleep-wake-driven influences on gene expression. The model reliably captured a wide range of dynamics in cortex, liver, and blood transcriptomes taken from mice and humans under various experimental conditions. Sleep-wake-driven factors outweighed circadian factors in driving gene expression in the cortex, whereas the opposite was observed in the liver and blood. Because of tissue- and gene-specific responses, sleep deprivation led to a long-lasting intra- and inter-tissue desynchronization. The model showed that recovery sleep contributed to these long-lasting changes. The results demonstrate that the analyses of the daily rhythms in gene expression must take the complex interactions between circadian and sleep-wake influences into account. A record of this paper's transparent peer review process is included in the supplemental information.
100
S. Bassani, J. Chrast, G. Ambrosini, N. Voisin, F. Schütz, A. Brusco, F. Sirchia, L. Turban, S. Schubert, R. Abou Jamra, J.U. Schlump, D. DeMille, P. Bayrak-Toydemir, G.R. Nelson, K.N. Wong, L. Duncan, M. Mosera, C. Gilissen, L.E.L.M. Vissers, R. Pfundt, R. Kersseboom, H. Yttervik, G.&.#.x.c.5.;.M. Hansen, M.F. Smeland, K.M. Butler, M.J. Lyons, C.M.B. Carvalho, C. Zhang, J.R. Lupski, L. Potocki, L. Flores-Gallegos, R. Morales-Toquero, F. Petit, B. Yalcin, A. Tuttle, H.Z. Elloumi, L. McCormick, M. Kukolich, O. Klaas, J. Horvath, M. Scala, M. Iacomino, F. Operto, F. Zara, K. Writzl, A. Maver, M.K. Haanpää, P. Pohjola, H. Arikka, A.J.A. Kievit, C. Calandrini, C. Iseli, N. Guex, A. Reymond
S Bassani et al.
Variant-specific pathophysiological mechanisms of AFF3 differently influence transcriptome profiles.
2024
Genome medicine
38,811,945
22/01/2025 11:12
30/05/2024 00:00
Bassani
12
16
1
We previously described the KINSSHIP syndrome, an autosomal dominant disorder associated with intellectual disability (ID), mesomelic dysplasia and horseshoe kidney, caused by de novo variants in the degron of AFF3. Mouse knock-ins and overexpression in zebrafish provided evidence for a dominant-negative mode of action, wherein an increased level of AFF3 resulted in pathological effects.
101
S. Bibert, M. Quinodoz, S. Perriot, F.S. Krebs, M. Jan, R.C. Malta, E. Collinet, M. Canales, A. Mathias, N. Faignart, E. Roulet-Perez, P. Meylan, R. Brouillet, O. Opota, L. Lozano-Calderon, F. Fellmann, N. Guex, V. Zoete, S. Asner, C. Rivolta, R. Du Pasquier, P.Y. Bochud
S Bibert et al.
Herpes simplex encephalitis due to a mutation in an E3 ubiquitin ligase.
2024
Nature communications
38,730,242
22/01/2025 11:12
10/05/2024 00:00
Bibert
60
15
1
Encephalitis is a rare and potentially fatal manifestation of herpes simplex type 1 infection. Following genome-wide genetic analyses, we identified a previously uncharacterized and very rare heterozygous variant in the E3 ubiquitin ligase WWP2, in a 14-month-old girl with herpes simplex encephalitis. The p.R841H variant (NM_007014.4:c.2522G > A) impaired TLR3 mediated signaling in inducible pluripotent stem cells-derived neural precursor cells and neurons; cells bearing this mutation were also more susceptible to HSV-1 infection compared to control cells. The p.R841H variant increased TRIF ubiquitination in vitro. Antiviral immunity was rescued following the correction of p.R841H by CRISPR-Cas9 technology. Moreover, the introduction of p.R841H in wild type cells reduced such immunity, suggesting that this mutation is linked to the observed phenotypes.
102
A. Brümmer, S. Bergmann
A Brümmer et al.
Disentangling genetic effects on transcriptional and post-transcriptional gene regulation through integrating exon and intron expression QTLs.
2024
Nature communications
38,710,690
22/01/2025 11:12
06/05/2024 00:00
Brümmer
60
15
1
Expression quantitative trait loci (eQTL) studies typically consider exon expression of genes and discard intronic RNA sequencing reads despite their information on RNA metabolism. Here, we quantify genetic effects on exon and intron levels of genes and their ratio in lymphoblastoid cell lines, revealing thousands of cis-QTLs of each type. While genetic effects are often shared between cis-QTL types, 7814 (47%) are not detected as top cis-QTLs at exon levels. We show that exon levels preferentially capture genetic effects on transcriptional regulation, while exon-intron-ratios better detect those on co- and post-transcriptional processes. Considering all cis-QTL types substantially increases (by 71%) the number of colocalizing variants identified by genome-wide association studies (GWAS). It further allows dissecting the potential gene regulatory processes underlying GWAS associations, suggesting comparable contributions by transcriptional (50%) and co- and post-transcriptional regulation (46%) to complex traits. Overall, integrating intronic RNA sequencing reads in eQTL studies expands our understanding of genetic effects on gene regulatory processes.
103
A. Schäfer, S.M. D'Almeida, J. Dorier, N. Guex, J. Villard, M. Garcia
A Schäfer et al.
Comparative assessment of cytometry by time-of-flight and full spectral flow cytometry based on a 33-color antibody panel.
2024
Journal of immunological methods
38,365,120
22/01/2025 11:12
01/04/2024 00:00
Schäfer
418
527
Mass cytometry and full spectrum flow cytometry have recently emerged as new promising single cell proteomic analysis tools that can be exploited to decipher the extensive diversity of immune cell repertoires and their implication in human diseases. In this study, we evaluated the performance of mass cytometry against full spectrum flow cytometry using an identical 33-color antibody panel on four healthy individuals. Our data revealed an overall high concordance in the quantification of major immune cell populations between the two platforms using a semi-automated clustering approach. We further showed a strong correlation of cluster assignment when comparing manual and automated clustering. Both comparisons revealed minor disagreements in the quantification and assignment of rare cell subpopulations. Our study showed that both single cell proteomic technologies generate highly overlapping results and substantiate that the choice of technology is not a primary factor for successful biological assessment of cell profiles but must be considered in a broader design framework of clinical studies.
104
K. Pinjusic, G. Ambrosini, J. Lourenco, N. Fournier, C. Iseli, N. Guex, O. Egorova, S. Nassiri, D.B. Constam
K Pinjusic et al.
Inhibition of anti-tumor immunity by melanoma cell-derived Activin-A depends on STING.
2023
Frontiers in immunology
38,304,252
22/01/2025 11:12
01/01/2023 00:00
Pinjusic
402
14
The transforming growth factor-β (TGF-β) family member activin A (hereafter Activin-A) is overexpressed in many cancer types, often correlating with cancer-associated cachexia and poor prognosis. Activin-A secretion by melanoma cells indirectly impedes CD8+ T cell-mediated anti-tumor immunity and promotes resistance to immunotherapies, even though Activin-A can be proinflammatory in other contexts. To identify underlying mechanisms, we here analyzed the effect of Activin-A on syngeneic grafts of Braf mutant YUMM3.3 mouse melanoma cells and on their microenvironment using single-cell RNA sequencing. We found that the Activin-A-induced immune evasion was accompanied by a proinflammatory interferon signature across multiple cell types, and that the associated increase in tumor growth depended at least in part on pernicious STING activity within the melanoma cells. Besides corroborating a role for proinflammatory signals in facilitating immune evasion, our results suggest that STING holds considerable potential as a therapeutic target to mitigate tumor-promoting Activin-A signaling at least in melanoma.
105
S. Bassani, J. Chrast, G. Ambrosini, N. Voisin, F. Schütz, A. Brusco, F. Sirchia, L. Turban, S. Schubert, R.A. Jamra, J.U. Schlump, D. DeMille, P. Bayrak-Toydemir, G.R. Nelson, K.N. Wong, L. Duncan, M. Mosera, C. Gilissen, L.E.L.M. Vissers, R. Pfundt, R. Kersseboom, H. Yttervik, G.&.#.x.c.5.;.M. Hansen, M. Falkenberg Smeland, K.M. Butler, M.J. Lyons, C.M.B. Carvalho, C. Zhang, J.R. Lupski, L. Potocki, L. Flores-Gallegos, R. Morales-Toquero, F. Petit, B. Yalcin, A. Tuttle, H.Z. Elloumi, L. Mccormick, M. Kukolich, O. Klaas, J. Horvath, M. Scala, M. Iacomino, F. Operto, F. Zara, K. Writzl, A. Maver, M.K. Haanpää, P. Pohjola, H. Arikka, C. Iseli, N. Guex, A. Reymond
S Bassani et al.
Variant-specific pathophysiological mechanisms of AFF3 differently influence transcriptome profiles.
2024
medRxiv : the preprint server for health sciences
38,293,053
22/01/2025 11:12
17/01/2024 00:00
Bassani
419
We previously described the KINSSHIP syndrome, an autosomal dominant disorder associated with intellectual disability (ID), mesomelic dysplasia and horseshoe kidney,caused by de novo variants in the degron of AFF3. Mouse knock-ins and overexpression in zebrafish provided evidence for a dominant-negative (DN) mode-of-action, wherein an increased level of AFF3 resulted in pathological effects.
106
M. Furrer, S.A. Meier, M. Jan, P. Franken, M.A. Sundset, S.A. Brown, G.C. Wagner, R. Huber
M Furrer et al.
Reindeer in the Arctic reduce sleep need during rumination.
2024
Current biology : CB
38,141,616
22/01/2025 11:12
22/01/2024 00:00
Furrer
353
34
2
Timing and quantity of sleep depend on a circadian (∼24-h) rhythm and a specific sleep requirement.1 Sleep curtailment results in a homeostatic rebound of more and deeper sleep, the latter reflected in increased electroencephalographic (EEG) slow-wave activity (SWA) during non-rapid eye movement (NREM) sleep.2 Circadian rhythms are synchronized by the light-dark cycle but persist under constant conditions.3,4,5 Strikingly, arctic reindeer behavior is arrhythmic during the solstices.6 Moreover, the Arctic's extreme seasonal environmental changes cause large variations in overall activity and food intake.7 We hypothesized that the maintenance of optimal functioning under these extremely fluctuating conditions would require adaptations not only in daily activity patterns but also in the homeostatic regulation of sleep. We studied sleep using non-invasive EEG in four Eurasian tundra reindeer (Rangifer tarandus tarandus) in Tromsø, Norway (69°N) during the fall equinox and both solstices. As expected, sleep-wake rhythms paralleled daily activity distribution, and sleep deprivation resulted in a homeostatic rebound in all seasons. Yet, these sleep rebounds were smaller in summer and fall than in winter. Surprisingly, SWA decreased not only during NREM sleep but also during rumination. Quantitative modeling revealed that sleep pressure decayed at similar rates during the two behavioral states. Finally, reindeer spent less time in NREM sleep the more they ruminated. These results suggest that they can sleep during rumination. The ability to reduce sleep need during rumination-undisturbed phases for both sleep recovery and digestion-might allow for near-constant feeding in the arctic summer.
107
G. Snieckute, L. Ryder, A.C. Vind, Z. Wu, F.S. Arendrup, M. Stoneley, S. Chamois, A. Martinez-Val, M. Leleu, R. Dreos, A. Russell, D.M. Gay, A.V. Genzor, B.S. Choi, A.L. Basse, F. Sass, M. Dall, L.C.M. Dollet, M. Blasius, A.E. Willis, A.H. Lund, J.T. Treebak, J.V. Olsen, S.S. Poulsen, M.E. Pownall, B.A.H. Jensen, C. Clemmensen, Z. Gerhart-Hines, D. Gatfield, S. Bekker-Jensen
G Snieckute et al.
ROS-induced ribosome impairment underlies ZAKα-mediated metabolic decline in obesity and aging.
2023
Science (New York, N.Y.)
38,060,659
22/01/2025 11:12
08/12/2023 00:00
Snieckute
17
382
6675
The ribotoxic stress response (RSR) is a signaling pathway in which the p38- and c-Jun N-terminal kinase (JNK)-activating mitogen-activated protein kinase kinase kinase (MAP3K) ZAKα senses stalling and/or collision of ribosomes. Here, we show that reactive oxygen species (ROS)-generating agents trigger ribosomal impairment and ZAKα activation. Conversely, zebrafish larvae deficient for ZAKα are protected from ROS-induced pathology. Livers of mice fed a ROS-generating diet exhibit ZAKα-activating changes in ribosomal elongation dynamics. Highlighting a role for the RSR in metabolic regulation, ZAK-knockout mice are protected from developing high-fat high-sugar (HFHS) diet-induced blood glucose intolerance and liver steatosis. Finally, ZAK ablation slows animals from developing the hallmarks of metabolic aging. Our work highlights ROS-induced ribosomal impairment as a physiological activation signal for ZAKα that underlies metabolic adaptation in obesity and aging.
108
A.N. Glaus, M. Brechet, G. Swinnen, L. Lebeigle, J. Iwaszkiewicz, G. Ambrosini, I. Julca, J. Zhang, R. Roberts, C. Iseli, N. Guex, J. Jiménez-Gómez, N. Glover, G.B. Martin, S. Strickler, S. Soyk
AN Glaus et al.
Repairing a deleterious domestication variant in a floral regulator gene of tomato by base editing.
2025
Nature genetics
39,747,596
22/01/2025 11:13
01/01/2025 00:00
Glaus
46
57
1
Crop genomes accumulate deleterious mutations-a phenomenon known as the cost of domestication. Precision genome editing has been proposed to eliminate such potentially harmful mutations; however, experimental demonstration is lacking. Here we identified a deleterious mutation in the tomato transcription factor SUPPRESSOR OF SP2 (SSP2), which became prevalent in the domesticated germplasm and diminished DNA binding to genome-wide targets. We found that the action of SSP2 is partially redundant with that of its paralog SSP in regulating shoot and inflorescence architecture. However, redundancy was compromised during tomato domestication and lost completely in the closely related species Physalis grisea, in which a single ortholog regulates shoot branching. We applied base editing to directly repair the deleterious mutation in cultivated tomato and obtained plants with compact growth that provide an early fruit yield. Our work shows how deleterious variants have sensitized modern genotypes for phenotypic tuning and illustrates how repairing deleterious mutations with genome editing may allow predictable crop improvement.
109
F. Brozzi, C. Jacovetti, C. Cosentino, V. Menoud, K. Wu, M.B. Bayazit, B. Abdulkarim, C. Iseli, N. Guex, C. Guay, R. Regazzi
F Brozzi et al.
tRNA-derived fragments in T lymphocyte-beta cell crosstalk and in type 1 diabetes pathogenesis in NOD mice.
2024
Diabetologia
38,967,669
22/01/2025 11:13
01/10/2024 00:00
Brozzi
420
67
10
tRNAs play a central role in protein synthesis. Besides this canonical function, they were recently found to generate non-coding RNA fragments (tRFs) regulating different cellular activities. The aim of this study was to assess the involvement of tRFs in the crosstalk between immune cells and beta cells and to investigate their contribution to the development of type 1 diabetes.
119
A.L. Ducrest, L.M. San-Jose, S. Neuenschwander, E. Schmid-Siegert, C. Simon, M. Pagni, C. Iseli, H. Richter, N. Guex, T. Cumer, E. Beaudoing, M. Dupasquier, P. Charruau, P. Ducouret, I. Xenarios, J. Goudet, A. Roulin
AL Ducrest et al.
Melanin and Neurotransmitter Signalling Genes Are Differentially Co-Expressed in Growing Feathers of White and Rufous Barn Owls.
2025
Pigment cell & melanoma research
39,910,963
06/02/2025 11:35
01/03/2025 00:00
Ducrest
421
38
2
Regulation of melanin-based pigmentation is complex, involving multiple genes. Because different genes can contribute to the same pigmentation phenotype, the genes identified in model organisms may not necessarily apply to wild species. In the barn owl (Tyto alba), ventral plumage colour ranges from white to rufous, with genetic variation in the melanocortin 1 receptor gene (MC1R) accounting for at least a third of this variation. In the present study, we used transcriptomic data to compare the gene expression profiles of growing feathers from nestlings with different MC1R genotypes. We identified 21 differentially expressed genes, nine of which are involved in melanogenesis, while seven are related to neurotransmitter function or synaptic activity. With the exception of CALB1, all of the differentially expressed genes were upregulated in rufous owls compared to white barn owls. To the best of our knowledge, this study is the first to link melanin production with neurotransmitter-related genes, and we discuss possible evolutionary explanations for this connection.
120
F. Mattioli, R. Friðriksdóttir, A. Hebert, S. Bassani, N. Ibrahim, S. Naz, J. Chrast, C. Pailler-Pradeau, &.#.x.c.1.;. Oddsson, P. Sulem, G.H. Halldorsson, P. Melsted, D.F. Guðbjartsson, F. Palombo, T. Pippucci, N. Nouri, M. Seri, E.G. Farrow, C.J. Saunders, N. Guex, M. Ansar, K. Stefansson, A. Reymond
F Mattioli et al.
Bi-allelic variants in BRF2 are associated with perinatal death and craniofacial anomalies.
2025
Genome medicine
40,229,899
15/04/2025 10:35
14/04/2025 00:00
Mattioli
12
17
1
Variants in genes encoding multiple subunits of the RNA Polymerase III complex which synthesizes rRNAs, tRNAs, and other small RNAs were previously associated with neurological disorders, such as syndromic hypomyelination leukodystrophies, pontocerebellar hypoplasia, and cerebellofaciodental syndrome. One new such candidate is BRF2, which encodes a TFIIB-like factor that recruits the RNA polymerase III complex to type 3 promoters to initiate transcription of U6, RnaseP, and 7SK RNAs.
121
M. Mouginot, S. Hani, P. Cousin, J. Dorier, A. Ravera, M.C. Gambetta
M Mouginot et al.
A boundary-defining protein facilitates megabase-scale regulatory chromosomal loop formation in Drosophila neurons.
2025
Genes & development
40,240,144
17/04/2025 10:35
02/06/2025 00:00
Mouginot
13
39
11-12
Regulatory elements, such as enhancers and silencers, control transcription by establishing physical proximity to target gene promoters. Neurons in flies and mammals exhibit long-range three-dimensional genome contacts, proposed to connect genes with distal regulatory elements. However, the relevance of these contacts for neuronal gene transcription and the mechanisms underlying their specificity necessitate further investigation. Here, we precisely disrupt several long-range contacts in fly neurons, demonstrating their importance for megabase-range gene regulation and uncovering a hierarchical process in their formation. We further reveal an essential role for the chromosomal boundary-forming protein Cp190 in anchoring many long-range contacts, highlighting a mechanistic interplay between boundary and loop formation. Finally, we develop an unbiased proteomics-based method to systematically identify factors required for specific long-range contacts. Our findings underscore the essential role of architectural proteins such as Cp190 in cell type-specific genome organization in enabling specialized neuronal transcriptional programs.
122
K.E. Carstens, E. Gronskaya, D. Jäckel, J. Bertoli, K.R. Cuevas, J. Dorier, S. Wang, D. Lopez-Rodriguez, T.J. Shafer, M.G. Zurich, D. Pamies
KE Carstens et al.
Application of a high-density microelectrode array assay using a 3D human iPSC-derived brain microphysiological system model for in vitro neurotoxicity screening of environmental compounds.
2025
Archives of toxicology
40,293,475
29/04/2025 10:35
01/07/2025 00:00
Carstens
422
99
7
Unraveling the associations between human exposure to environmental chemicals and potential neurotoxicity presents significant challenges. Evaluation of neurotoxicity potential using animal testing is resource-intensive (financial, labor, and animal use) and faces uncertainties regarding biological relevance to human health outcomes. Therefore, there is a need to develop efficient and human-relevant in vitro new approach methodologies (NAMs) to screen and evaluate chemicals for neurotoxicity potential. Recording of neural network activity using microelectrode array (MEA) technology has been identified as a reliable and reproducible method for evaluating neurotoxicity. Much of this research has been performed in 2D rodent-derived cell models. The 'BrainSpheres MEA assay' described in this study offers a promising functional human induced pluripotent stem cell (iPSC)-derived 3D brain model comprising neurons, astrocytes, and oligodendrocytes. We demonstrate consistent spontaneous neuronal firing and network bursting parameters from 7-week-old BrainSpheres using a high-density MEA technology. The performance of this model as a human-relevant NAM was evaluated by conducting a multi-concentration, 13 day exposure study with a set of ten chemicals. Neural activity metrics were assessed and compared to results from a 2D-MEA assay using rodent cells. Loperamide and domoic acid (two assay positive controls) demonstrated similar bioactivity profiles in the BrainSphere MEA assay to the 2D-MEA assay, while acetaminophen (assay negative control) was inactive in both assays. The 2D-MEA model demonstrated more potent bioactivity for 4/7 chemicals that were active in both assays. In the future, reducing replicate variability and testing a larger set of chemicals will likely improve the accuracy and reliability of the assay. These preliminary findings suggest that the BrainSphere assay could be used alongside the rat network formation assay (rNFA) as part of a tiered strategy, where hits in the rNFA are confirmed and further characterized in the BrainSphere model, helping move toward animal-free toxicological testing.
125
C. Ronchi, G. Ambrosini, F. Hughes, R.L. Flaherty, H.M. Quinn, D. Matvienko, A. Agnoletto, C. Brisken
C Ronchi et al.
biogrowleR: Enhancing Longitudinal Data Analysis.
2025
Journal of mammary gland biology and neoplasia
40,459,798
04/06/2025 10:35
03/06/2025 00:00
Ronchi
423
30
1
Time course measurements are used for many applications in biomedical research, ranging from growth curves to drug efficacy testing and high-throughput screening. Statistical methods used to analyze the resulting longitudinal data, such as t-tests or repeated measures ANOVA have limitations when groups are unbalanced, or individual measurements are missing. To address these issues we developed biogrowleR (https://upbri.gitlab.io/biogrowleR/), a workflow to visualize and analyze data based on Frequentist and Bayesian inference combined with hierarchical modeling. By focusing on effect sizes we enhance data interpretation. The workflow further includes a randomization algorithm important to reduce numbers of experimental animals (RRR) and costs. The workflow and R package were designed to be used by researchers with limited experience in R and biostatistics. Our open-source R package biogrowleR contains tutorials, pipelines, and helper functions for the analysis of longitudinal data and enables non computational scientists to perform more effective data analysis.
128
E. Ghisoni, F. Benedetti, A. Minasyan, M. Desbuisson, P. Cunnea, A.J. Grimm, N. Fahr, C. Capt, N. Rayroux, F. De Carlo, D.C. Gulhan, J. Dagher, D. Barras, M. Morotti, J.A. Marín-Jiménez, B.S. Chap, T. Santoro, G. Spagnol, M. Fleury, K. Fortis, J. Dorier, M.K. Townsend, S. Tissot, S. Rusakiewicz, H.J. Ferreira, A.I. Kraemer, M. Bassani-Stenberg, E.M. Swisher, L.A. Kandalaft, S.A. Mastroyannis, K.T. Montone, D.J. Powell, S. Banerjee, K.L. Terry, S.S. Tworoger, M.J. Pittet, J.L. Tanyi, G. Coukos, M.A. Merritt, C. Fotopoulou, J.R. Conejo-Garcia, D.D. Laniti
E Ghisoni et al.
Myeloid cell networks govern re-establishment of original immune landscapes in recurrent ovarian cancer.
2025
Cancer cell
40,749,672
03/08/2025 10:35
11/08/2025 00:00
Ghisoni
424
43
8
Immunotherapy has shown limited success in recurrent ovarian cancer (OC), with prognostic insights largely derived from treatment-naive tumors. We analyzed 697 tumor samples (566 primary and 131 recurrent) from 595 OC patients across five independent cohorts, capturing tumor-infiltrating lymphocytes (TILs) heterogeneity and identifying four immune phenotypes linked to prognosis and TIL:myeloid networks driving malignant progression. We found that in preclinical mouse models, mirroring inflamed human OCs, the recurrent Brca1mut tumors maintained activated TILs:dendritic cells (DCs) niches but evaded immune control through upregulation of COX/PGE2 signaling. Conversely, recurrent Brca1wt tumors displayed loss of TILs:DCs niches and accumulated immunosuppressive tumor microenvironment (TME) networks featuring Trem2/ApoEhigh tumor associated macrophages (TAMs) and Nduf4l2high/Galectin3high malignant states. Recurrent tumors recapitulate the immunogenic landscapes of original cancers. Our findings reveal BRCA-dependent TIL:myeloid crosstalk as key to persistent immunogenicity in recurrent OC and propose new targets to enhance chemotherapy efficacy.
130
N. Eling, J. Dorier, S. Rusakiewicz, R. Liechti, P. Devanand, M. Daniel, J. Windhager, B.P. Fernandez, S. Déglise, L. Despland, A. Benyagoub, M. Możejko, D. Uchal, E. Szczurek, A. Loboda, D. Sandkuijl, N. Parsotam, H.S. Hong, M. Morfouace, N. Guex, G. Coukos, B. Bodenmiller, S. Tissot, D. Schulz
N Eling et al.
Multi-modal image analysis for large-scale cancer tissue studies within IMMUcan.
2025
Cell reports methods
40,930,089
11/09/2025 10:35
15/09/2025 00:00
Eling
425
5
9
In cancer research, multiplexed imaging allows detailed characterization of the tumor microenvironment (TME) and its link to patient prognosis. The integrated immunoprofiling of large adaptive cancer patient cohorts (IMMUcan) consortium collects multi-modal imaging data from thousands of patients with cancer to perform broad molecular and cellular spatial profiling. Here, we describe and compare two workflows for multiplexed immunofluorescence (mIF) and imaging mass cytometry (IMC) developed within IMMUcan to enable the generation of standardized data for cancer tissue analysis. The IFQuant software supports web-based, user-friendly, and reproducible analysis of mIF data. High sample throughput for IMC is achieved by optimizing experimental protocols, developing a robotic arm for automated slide loading, and classification-based cell typing. Using our manually labeled single-cell data, we show that tree-based methods outperform other cell-phenotyping tools. These pipelines form the basis for multiplexed image analysis within IMMUcan, and we summarize our learnings from 5 years of development and optimization.
133
D.M. Coda, L. Watt, L. Glauser, M.Y. Batiuk, A.M. Burns, C.L. Stahl, L.Y. Wong, J. Gräff
DM Coda et al.
Cell-type- and locus-specific epigenetic editing of memory expression.
2025
Nature genetics
41,162,785
30/10/2025 11:35
01/11/2025 00:00
Coda
46
57
11
Epigenetic mechanisms have long been proposed to act as molecular mnemonics1-3, but whether the epigenetic makeup of a single genomic site can guide learnt behaviors remains unknown. Here we combined CRISPR-based epigenetic editing tools4,5 with c-Fos-driven engram technologies6,7 to address this question in memory-bearing neuronal ensembles. Focusing on the promoter of Arc, which encodes a master regulator of synaptic plasticity8, we found that its locus-specific and temporally controlled epigenetic editing is necessary and sufficient to regulate memory expression. Such effects occurred irrespective of the memory phase-during the initially labile period after learning and for fully consolidated memories-and were reversible within subject, testifying to their inherent plasticity. These findings provide a proof-of-principle that site-specific epigenetic dynamics are causally implicated in memory expression.
134
I.E. Vorontsov, I. Kozin, S. Abramov, A. Boytsov, A. Jolma, M. Albu, G. Ambrosini, K. Faltejskova, A.J. Gralak, N. Gryzunov, S. Inukai, S. Kolmykov, P. Kravchenko, J.F. Kribelbauer-Swietek, K.U. Laverty, V. Nozdrin, Z.M. Patel, D. Penzar, M.L. Plescher, S.E. Pour, R. Razavi, A.W.H. Yang, I. Yevshin, A. Zinkevich, M.T. Weirauch, P. Bucher, B. Deplancke, O. Fornes, J. Grau, I. Grosse, F.A. Kolpakov, , V.J. Makeev, T.R. Hughes, I.V. Kulakovskiy
IE Vorontsov et al.
Cross-platform motif discovery and benchmarking to explore binding specificities of poorly studied human transcription factors.
2025
Communications biology
41,203,846
08/11/2025 11:35
07/11/2025 00:00
Vorontsov
171
8
1
A sequence motif representing the DNA-binding specificity of a transcription factor (TF) is commonly modelled with a positional weight matrix (PWM). Focusing on understudied human TFs, we processed results of 4,237 experiments for 394 TFs, assayed using five different experimental platforms. By human curation, we approved a subset of experiments that yielded consistent motifs across platforms and replicates, and evaluated quantitatively the cross-platform performance of PWMs obtained with ten motif discovery tools. Notably, nucleotide composition and information content are not correlated with motif performance and do not help in detecting underperformers, while motifs with low information content, in many cases, describe well the binding specificity assessed across different experimental platforms. By combining multiple PMWs into a random forest, we demonstrate the potential of accounting for multiple modes of TF binding. Finally, we present the Codebook Motif Explorer ( https://mex.autosome.org ), cataloguing motifs, benchmarking results, and the underlying experimental data.
135
E. Skoufa, J. Zhong, K. Hu, O. Kahre, G. Tsissios, L. Carrau, A. Herrera, A. Dominguez Mantes, M. Leleu, A. Castilla-Ibeas, H. Jang, M. Weigert, G. La Manno, M. Lutolf, M. Ros, C. Aztekin
E Skoufa et al.
Specialized signaling centers direct cell fate and spatial organization in a mesodermal organoid model.
2025
Science advances
41,313,763
29/11/2025 11:35
28/11/2025 00:00
Skoufa
80
11
48
Specialized signaling centers orchestrate robust development and regeneration. Limb morphogenesis, for instance, requires interactions between the mesoderm and the signaling center apical-ectodermal ridge (AER), whose properties and role in cell fate decisions have remained challenging to dissect. To tackle this, we developed mouse embryonic stem cell (mESC)-based heterogeneous cultures and a three-dimensional (3D) organoid model, termed budoids, comprising cells with AER, surface ectoderm, and mesoderm properties. mESCs were first induced into heterogeneous cultures that self-organized into domes in 2D. Aggregating these cultures formed mesodermal organoids with certain limb bud-like features in 3D, exhibiting chondrogenesis-based symmetry breaking and elongation. Using our organoids and quantitative in situ expression profiling, we uncovered that AER-like cells support nearby limb mesoderm and fibroblast identities while enhancing tissue polarization that permits distant cartilage formation. Together, our findings provide a powerful model to study epithelial signaling center-mesoderm interactions during morphogenesis and reveal the ability of signaling center AER cells to concurrently modulate cell fate and spatial organization.
136
S. Paulišić, A. Boccaccini, R. Dreos, G. Ambrosini, N. Guex, R.M. Benstein, M. Schmid, C. Fankhauser
S Paulišić et al.
Transcriptional dynamics and chromatin accessibility in the regulation of shade-responsive genes in Arabidopsis.
2025
Genome biology
41,372,949
11/12/2025 11:36
10/12/2025 00:00
Paulišić
10
26
1
Open chromatin regions host DNA regulatory motifs that are accessible to transcription factors and the transcriptional machinery. In Arabidopsis, responses to light are heavily regulated at the transcriptional level. Shade, for example, can limit photosynthesis and is rapidly perceived by phytochromes as a reduction of red to far-red light ratio (LRFR). Under shade, phytochromes become inactive, enabling PHYTOCHROME INTERACTING FACTORs (PIFs), particularly PIF7, to promote genome-wide reprogramming essential for LRFR responses. An initial strong and fast regulation of shade-responsive genes is followed by attenuation of this response under prolonged shade.
140
G. Paduthol, M. Nikolaev, K. Sharma, J. Blanc, K. Tomasek, L.I.E. Schlunke, V. Borgeat, G. Ambrosini, I. Kolotuev, S. Clerc-Rosset, N. Gjorevski, G.W. Knott, M.P. Lütolf, V.V. Thacker, J.D. McKinney
G Paduthol et al.
A microphysiological human mini-bladder reveals urine-urothelium interplay in tissue resilience and UPEC recurrence in urinary tract infections.
2026
Nature communications
41,634,008
04/02/2026 11:36
03/02/2026 00:00
Paduthol
60
17
1
Urine is a dynamic and highly variable biofluid. Urine-urothelium interactions are a critical yet underexplored factor in bladder homoeostasis and urinary tract infections (UTIs). Here, we report on a human 'mini-bladder' model that exposes a stratified urothelium to urine of defined composition, and incorporates micturition. Prolonged exposure to high-solute concentration urine weakens tight junctions, dysregulates immune responses, and reduces bladder tissue resilience. This increases susceptibility to colonisation of the bladder by uropathogenic Escherichia coli (UPEC) which reduces efficacy of antibiotic therapy. In high-solute concentration urine, Fosfomycin monotherapy - prescribed for uncomplicated UTIs, induces the formation of cell wall-deficient (CWD) UPEC in the urine (as observed in patients with recurrent UTIs) but also within deeper urothelial layers. Tissue-associated CWD UPEC directly contributes to recurrence. Our findings expand the conceptual role for CWD UPEC in UTIs, and demonstrate the power of the mini-bladder platform to capture urine-urothelial microenvironment dynamics that actively shape UTI pathogenesis and antibiotic tolerance.
id
Authors
authors
Title
Year
Journal
pmid
created_at
doi
published_at
first_author
journal_id
user_id
volume
issue
abstract
Link to paper
40
J. den Hoed, E. de Boer, N. Voisin, A.J.M. Dingemans, N. Guex, L. Wiel, C. Nellaker, S.M. Amudhavalli, S. Banka, F.S. Bena, B. Ben-Zeev, V.R. Bonagura, A.-.L. Bruel, T. Brunet, H.G. Brunner, H.B. Chew, J. Chrast, L. Cimbalistienė, H. Coon, E.C. Délot, F. Démurger, A.-.S. Denommé-Pichon, C. Depienne, D. Donnai, D.A. Dyment, O. Elpeleg, L. Faivre, C. Gilissen, L. Granger, B. Haber, Y. Hachiya, Y. .H.a.m.z.a.v.i. Abedi, J. Hanebeck, J.Y. Hehir-Kwa, B. Horist, T. Itai, A. Jackson, R. Jewell, K.L. Jones, S. Joss, H. Kashii, M. Kato, A.A. Kattentidt-Mouravieva, F. Kok, U. Kotzaeridou, V. Krishnamurthy, V. Kučinskas, A. Kuechler, A. Lavillaureix, P. Liu, L. Manwaring, N. Matsumoto, B. Mazel, K. McWalter, V. Meiner, M.A. Mikati, S. Miyatake, T. Mizuguchi, L.H. Moey, S. Mohammed, H. Mor-Shaked, H. Mountford, R. Newbury-Ecob, S. Odent, L. Orec, M. Osmond, T.B. Palculict, M. Parker, A. Petersen, R. Pfundt, E. Preikšaitienė, K. Radtke, E. Ranza, J.A. Rosenfeld, T. Santiago-Sim, C. Schwager, M. Sinnema, L. .S.n.i.j.d.e.r.s. Blok, R.C. Spillmann, A.P.A. Stegmann, I. Thiffault, L. Tran, A. Vaknin-Dembinsky, J.H. Vedovato-dos-Santos, S.A. Vergano, E. Vilain, A. Vitobello, M. Wagner, A. Waheeb, M. Willing, B. Zuccarelli, U. Kini, D.F. Newbury, T. Kleefstra, A. Reymond, S.E. Fisher, L.E.L.M. Vissers
J den Hoed et al.
Mutation-specific pathophysiological mechanisms define different neurodevelopmental disorders associated with SATB1 dysfunction
2020
bioRxiv
17/12/2020 10:25
10.1101/2020.10.23.352278
24/10/2020 00:00
den Hoed
397
42
N. Bararpour, F. Gilardi, C. Carmeli, J. Sidibe, J. Ivanisevic, T. Caputo, M. Augsburger, S. Grabherr, B.é. Desvergne, N. Guex, M. Bochud, A. Thomas
N Bararpour et al.
Visualization and normalization of drift effect across batches in metabolome-wide association studies
2020
bioRxiv
17/12/2020 10:26
10.1101/2020.01.22.914051
22/01/2020 00:00
Bararpour
397
47
P. .G. Manti, F. Darbellay, M. Leleu, B. Moret, J. Cuennet, F. Droux, M. Stoudmann, G.-.F. Mancini, A.è. Hautier, Y. Barrandon
P G Manti et al.
Early mechanisms of whisker development: Prdm1 and its regulation in whisker development and evolutionary loss
2021
bioRxiv
13/03/2021 11:35
10.1101/2021.03.11.433122
12/03/2021 00:00
Manti
397
49
M. Graeff, S. Rana, J.o.s. .R. Wendrich, J. Dorier, T. Eekhout, A. .C. .A. Fandino, N. Guex, G.e.o.r.g.e. .W. Bassel, B. De Rybel, C.h.r.i.s.t.i.a.n. .S. Hardtke
M Graeff et al.
A morpho-transcriptomic map of brassinosteroid action in the Arabidopsis root
2021
bioRxiv
01/04/2021 10:35
10.1101/2021.03.30.437656
31/03/2021 00:00
Graeff
397
59
A. Seifinejad, A. Neiteler, S. Li, C. Pfister, R. Fronczek, L. Shan, L.e.e. .A. Garrett-Sinha, D. Frieser, M. Honda, Y. Arribat, D. Grepper, F. Amati, M. Picot, C. Iseli, N. Chartrel, G.e.r.t. .J. Lammers, R. Liblau, A. Vassalli, M. Tafti
A Seifinejad et al.
Narcolepsy with cataplexy is caused by epigenetic silencing of hypocretin neurons
2021
bioRxiv
22/10/2021 12:09
10.1101/2021.09.21.461046
24/09/2021 00:00
Seifinejad
397
61
N. Gobet, M. Jan, P. Franken, I. Xenarios
N Gobet et al.
Towards mouse genetic-specific RNA-sequencing read mapping
2021
bioRxiv
20/11/2021 11:35
10.1101/2021.10.01.462776
01/10/2021 00:00
Gobet
397
62
J.u.l.i.a.n. .C. Shillcock, J. Hastings, N. Riguet, H. Lashuel
Julian C Shillcock et al.
Non-monotonic fibril surface occlusion by GFP tags from coarse-grained molecular simulations
2021
bioRxiv
20/11/2021 11:35
10.1101/2021.10.21.465309
22/10/2021 00:00
Shillcock
397
65
V. Barquissau, N.è. Zanou, S. Geller, J. Castillo-Armengol, F. Marzetta, K. Huber, D. Ziegler, I. Lopez-Mejia, J. .B. Fernandez, C. Roger, N. Guex, F.é.é. Preitner, J.-.M. Vanacker, L. Fajas
V Barquissau et al.
CDK4 deletion in mice prevents fat accumulation and increases endurance capacity through activation of estrogen-related receptor (ERR)-driven oxidative metabolism in skeletal muscle
2022
bioRxiv
05/03/2022 11:35
10.1101/2022.03.03.482783
04/03/2022 00:00
Barquissau
397
66
H.i.l.a.l. .A. Lashuel, A.-.L. Mahul-Mellier, S. Novello, R. .N. Hegde, Y. Jasiqi, M. .F. Altay, S. Donzelli, S.e.a.n. .M. DeGuire, R. Burai, P. Magalhães, A. Chiki, J. Ricci, M. Boussouf, A. Sadek, E. Stoops, C. Iseli, N. Guex
Hilal A Lashuel et al.
Neighbouring modifications interfere with the detection of phosphorylated alpha-synuclein at Serine 129: Revisiting the specificity of pS129 antibodies
2022
bioRxiv
01/04/2022 10:35
10.1101/2022.03.30.486322
31/03/2022 00:00
Lashuel
397
67
G. Ambrosini, A. Agnoletto, C. Brisken, P. Bucher
G Ambrosini et al.
The Breast Cancer Epigenomics Track Hub
2022
bioRxiv
05/05/2022 10:35
10.1101/2022.05.01.490187
01/05/2022 00:00
Ambrosini
397
77
M. Ugolini, K. Kuznetsova, H. Oda, H. Kimura, N.a.d.i.n.e. .L. Vastenhouw
M Ugolini et al.
Transcription bodies regulate gene expression by sequestering CDK9
2022
bioRxiv
22/11/2022 11:35
10.1101/2022.11.21.517317
21/11/2022 00:00
Ugolini
397
79
N. Vesel, C. Iseli, N. Guex, A. Lemopoulos, M. Blokesch
N Vesel et al.
DNA modifications impact natural transformation of
Acinetobacter baumannii
2023
bioRxiv
11/02/2023 11:37
10.1101/2023.02.09.527895
09/02/2023 00:00
Vesel
397
85
M. Pirovino, C. Iseli, J.o.s.e.p.h. .A. Curran, B. Conrad
M Pirovino et al.
Biomathematical model of prebiotic autocatalytic RNA networks shows birth of adaptive immunity from degenerating molecular parasite catalysts
2023
bioRxiv
27/05/2023 10:36
10.1101/2023.05.25.542273
25/05/2023 00:00
Pirovino
397
86
V. Gardeux, R.o.e.l. .P.J. Bevers, F.a.b.r.i.c.e. .P.A. David, E. Rosschaert, R. Rochepeau, B. Deplancke
V Gardeux et al.
DGRPool: A web tool leveraging harmonized
Drosophila
Genetic Reference Panel phenotyping data for the study of complex traits
2023
bioRxiv
06/06/2023 10:36
10.1101/2023.06.01.543194
05/06/2023 00:00
Gardeux
397
88
M. Jan, S. Jimenez, C.h.a.r.l.o.t.t.e. .N. Hor, D.-.J. Dijk, A.n.n.e. .C. Skeldon, P. Franken
M Jan et al.
Model integration of circadian and sleep-wake driven contributions to rhythmic gene expression reveals novel regulatory principles
2023
bioRxiv
15/08/2023 10:36
10.1101/2023.08.10.552614
14/08/2023 00:00
Jan
397
110
N. Eling, J. Dorier, S. Rusakiewicz, R. Liechti, P. Devanand, M. Daniel, J. Windhager, B. .P. Fernandez, S. Déglise, L. Despland, A. Benyagoub, A. Loboda, D. Sandkuijl, N. Parsotam, H. .S. Hong, M. Morfouace, N. Guex, G. Coukos, B. Bodenmiller, S. Tissot, D. Schulz
N Eling et al.
Multi-modal image analysis for large scale cancer tissue studies within IMMUcan
2025
bioRxiv
23/01/2025 11:36
10.1101/2024.12.16.628597
03/01/2025 00:00
Eling
397
111
D. .M. Coda, L.I.S.A. Watt, glauser, M. Batiuk, A. Burns, C. Stahl, J. Graeff
D M Coda et al.
Cell type and locus-specific epigenetic editing of memory expression
2024
bioRxiv
23/01/2025 11:36
10.1101/2024.08.18.608454
18/08/2024 00:00
Coda
397
112
M. Pirovino, C. Iseli, J.o.s.e.p.h. .A. Curran, B. Conrad
M Pirovino et al.
Biomathematical enzyme kinetics model of prebiotic autocatalytic RNA networks: degenerating parasite-specific hyperparasite catalysts confer parasite resistance and herald the birth of molecular immunity
2024
bioRxiv
23/01/2025 11:36
10.1101/2024.05.14.594066
17/05/2024 00:00
Pirovino
397
113
S. Martin, F. Fournes, G. Ambrosini, C. Iseli, K. Bojkowska, J. Marquis, N. Guex, J. Collier
S Martin et al.
DNA methylation by CcrM contributes to genome maintenance in the
Agrobacterium tumefaciens
plant pathogen
2023
bioRxiv
23/01/2025 11:36
10.1101/2023.11.28.569018
28/11/2023 00:00
Martin
397
114
G. Tsissios, M. Leleu, K. Hu, A. .E. Demirtas, H. Hu, T. Kawanishi, E. Skoufa, A. Valente, A. Herrera, A. Mery, L. Noseda, H. Ochi, S. Sakar, M. Tanaka, F. Zenk, C. Aztekin
G Tsissios et al.
Species-specific oxygen sensing governs the initiation of vertebrate limb
regeneration
2024
bioRxiv
23/01/2025 11:36
10.1101/2024.12.19.629359
20/12/2024 00:00
Tsissios
397
115
R. Meurs, M. De Matos, A. Bothe, N. Guex, T. Weber, A.u.r.e.l.i.o. .A. Teleman, N. Ban, D. Gatfield
R Meurs et al.
An
in vitro
assay of MCTS1-DENR-dependent re-initiation and ribosome profiling uncover the activity of MCTS2 and distinct function of eIF2D
2024
bioRxiv
23/01/2025 11:36
10.1101/2024.06.05.597545
05/06/2024 00:00
Meurs
397
116
E. Ghisoni, F. Benedetti, A. Minasyan, P. Cunnea, A.l.i.z.é.e. .J. Grimm, N.é. Fahr, M. Desbuisson, C. Capt, N. Rayroux, D.o.g.a. .C. Gulhan, J. Dagher, D. Barras, M. Morotti, J.u.a.n. .A. Marín-Jiménez, F. De Carlo, B. .S. Chap, G. Spagnol, M. Fleury, K. Fortis, J. Dorier, S. Tissot, S. Rusakiewicz, H.u.m.b.e.r.t.o. .J. Ferreira, M. Bassani-Stenberg, E.l.i.z.a.b.e.t.h. .M. Swisher, L. Kandalft, S.p.y.r.i.d.o.n. .A. Mastroyannis, K.a.t.h.l.e.e.n. .T. Montone, D. Powell, M.i.k.a.ë.l. .J. Pittet, J.a.n.o.s. .L. Tanyi, G. Coukos, C. Fotopoulou, J.o.s.e. .R. Conejo-Garcia, D. .D. Laniti
E Ghisoni et al.
Myeloid cell networks determine reinstatement of original immune environments in recurrent ovarian cancer
2024
bioRxiv
23/01/2025 11:36
10.1101/2024.05.02.590528
05/05/2024 00:00
Ghisoni
397
117
G. Vizzarro, A. Lemopoulos, D. .W. Adams, M. Blokesch
G Vizzarro et al.
Vibrio cholerae
pathogenicity island 2 encodes two distinct types of restriction systems
2024
bioRxiv
23/01/2025 11:36
10.1101/2024.04.04.588119
04/04/2024 00:00
Vizzarro
397
118
M. Bérard, L. Merlini, S. .G. Martin
M Bérard et al.
TORC1 reactivation by pheromone signaling revealed by phosphoproteomics of fission yeast sexual reproduction
2024
bioRxiv
23/01/2025 11:36
10.1101/2024.06.04.597361
06/06/2024 00:00
Bérard
397
123
M. Stock, M. .G. Angel, M. Witzenberger, J. Nowak, A. Biela, A. Chramiec-Glabik, V. Mariano, R.é. Dreos, A.é. Sklias, M. Quadroni, A. Brümmer, H. Gallart-Ayala, J. Ivanisevic, N. Guex, L. Kleenman, S. Leidel, M. Helm, C. Bagni, S. Glatt, D. Gatfield, S. Schwartz, J.-.Y. Roignant
M Stock et al.
Pus7
mutation links tRNA dysregulation to aggressive behavior through activation of the integrated stress response and glycolytic reprogramming
2025
bioRxiv
16/05/2025 10:36
10.1101/2025.05.12.653498
12/05/2025 00:00
Stock
397
124
N. Martinez-Sanchez, A. Brümmer, N.i.c.h.o.l.a. .J. Barron, M. Voronkov, D.a.n.i.e.l. .B. Rosoff, A.é. Liechti, E.d.w.a.r.d. .A. Hayter, S.é. Chamois, R.é. Dreos, N. Guex, E. Johnson, M. Baxter, K.e.r.r.i. .L.M. Smith, R. .C. Northeast, G. Galli, L. Hodson, D. Gatfield, D.a.v.i.d. .W. Ray, D.a.v.i.d. .A. Bechtold
N Martinez-Sanchez et al.
Ribosomal modification by BUD23 drives selective translational control over energy state and metabolic health
2025
bioRxiv
20/05/2025 10:36
10.1101/2025.05.16.654455
19/05/2025 00:00
Martinez-Sanchez
397
126
J. Lemaitre, D.é.é. Popelka, B. Ribotta, H. Westlake, S. Chakrabarti, L. Xiaoxue, M.a.r.k. .A. Hanson, H. Jiang, F. Di Cara, E. Kurant, F. David, B. Lemaitre
J Lemaitre et al.
A retrospective analysis of 400 publications reveals patterns of irreproducibility across an entire life sciences research field
2025
bioRxiv
10/07/2025 10:36
10.1101/2025.07.07.663460
09/07/2025 00:00
Lemaitre
397
127
H. Westlake, F. David, Y. Tian, K. Krakovic, A. Dolgikh, L. Juravlev, T. Esmangart de Bournonville, A. Carboni, C. Melcarne, T. Shan, Y. Wang, Y. Mu, A. Kotwal, N. Pirko, J. .P. Boquete, F. Schüpfer, S. Rommelaere, M. Poidevin, Z. Liu, S. Kondo, G.i.r.i.s.h. .S. Ratnaparkhi, S. Chakrabarti, G. Liu, F. Masson, L. Xiaoxue, M.a.r.k. .A. Hanson, H. Jiang, F. Di Cara, E. Kurant, B. Lemaitre
H Westlake et al.
Reproducibility of Scientific Claims in
Drosophila
Immunity: A Retrospective Analysis of 400 Publications
2025
bioRxiv
11/07/2025 10:36
10.1101/2025.07.07.663442
09/07/2025 00:00
Westlake
397
129
R.e.n.é.e. .L. Flaherty, F. Hughes, G. Sflomos, C. Ronchi, H. Kemp, T. Roumeliotis, A.n.y.a. .A. Nicholas, G. Ambrosini, A. Ziehme, S. Becker, Y. Zhang, H.a.z.e.l. .M. Quinn, L. Battista, H. Padda, S. Pezot, S. Jouny, Y. Liu, R. Brough, R. Marlow, M. Iravani, A. Okines, N.i.c.h.o.l.a.s. .C. Turner, A. Stavrodimou, K. Zaman, M. Fiche, B.e.a.t.r.i.c.e. .A. Howard, J.y.o.t.i. .S. Choudhary, V. Sanz-Moreno, C.l.a.r.e. .M. Isacke, L. Perryman, W. Jarolimek, S. Haider, C.h.r.i.s.t.o.p.h.e.r. .J. Lord, C. Brisken
Renée L Flaherty et al.
LOX inhibition disrupts a collagen-integrin–MYC axis as a translatable targeting strategy in invasive lobular carcinoma
2025
bioRxiv
06/09/2025 10:36
10.1101/2025.08.27.672618
01/09/2025 00:00
Flaherty
397
131
A. Jolma, A. Hernandez-Corchado, A.l.l.y. .W.H. Yang, A. Fathi, K.a.i.t.l.i.n. .U. Laverty, A. Brechalov, R. Razavi, M. Albu, H. Zheng, I.v.a.n. .V. Kulakovskiy, H.a.m.e.d. .S. Najafabadi, T.i.m.o.t.h.y. .R. Hughes
A Jolma et al.
GHT-SELEX demonstrates unexpectedly high intrinsic sequence specificity and complex DNA binding of many human transcription factors
2024
bioRxiv
25/10/2025 10:36
10.1101/2024.11.11.618478
12/11/2024 00:00
Jolma
397
132
K. Tomasek, K. Skurvydaite, G. Paduthol, A.l.l.i.s.o.n. .M. Burns, L.é. Schlunke, C. Pasquali, M. Romani, J.o.h.n. .D. McKinney
K Tomasek et al.
Epithelial Reprogramming by OM-89 Enhances Antibiotic Clearance of Uropathogenic
E. coli
in a Bladder Organoid Model
2025
bioRxiv
25/10/2025 10:36
10.1101/2025.10.22.683857
22/10/2025 00:00
Tomasek
397
137
J.é.ô. Perrard, C. Guay, N.è. Zanou, R. Regazzi
Jéô Perrard et al.
The MIME-seq technique allows to monitor the interaction of small non-coding RNAs with Argonaute proteins and their transfer to other cells
2025
bioRxiv
20/01/2026 11:36
10.64898/2025.12.18.695118
18/12/2025 00:00
Perrard
397
138
S. Poddar, F. Brozzi, C. Cosentino, C.é. Jacovetti, C. Guay, J.é.ô. Perrard, R. Regazzi
S Poddar et al.
Role of small intronic RNAs in the crosstalk between immune cells and β-cells during type 1 diabetes development
2025
bioRxiv
20/01/2026 11:36
10.64898/2025.12.19.695414
19/12/2025 00:00
Poddar
397
139
J. Chrast, S.t.e.p.h.a.n. .C. Collins, C. Roger, S. Ben-Ayache, C. Auwerx, P. Rogg, F. Vacca, O.é. Musset, L. Gueneau, Y. Emmenegger, F. Allias, F. Tran-Mau-Them, D.a.n.i.e.l. .C. Koboldt, M.a.t.t.h.e.w. .T. Pastore, K.r.i.s.t.e.n. .V. Truxal, L.i.n.d.a. .Z. Rossetti, L. Walker, K. Sadrieh, M.a.t.t.h.e.w. .A. Deardorff, C. Olimpio, J. Rosenblum, M. Meuwissen, A.n.n.a. .C. Jansen, J. Hassell, M. Kurian, A. Perez Caballero, B. Paternoster, O. Klaas, A. Busche, J. Horvath, J.-.H. Caberg, S. Alkan, M.a.r.i.j.n. .F. Stokman, S. Baskin, N. Goss, H. Gallart-Ayala, M. Ansar, Z. Kutalik, J. Ivanisevic, N. Guex, B. Yalcin, A. Reymond
J Chrast et al.
Lipid transport is necessary for neocortical lamination